рефераты конспекты курсовые дипломные лекции шпоры

Реферат Курсовая Конспект
Курс лекций по общей химии
Курс лекций по общей химии - Лекция, раздел Химия, Казанский Государственный Технический Университет Им. А.н. Туполева ...
Казанский государственный технический университет им. А.Н. Туполева
Курс лекций по общей химии
Казань 2005
УДК 54
ББК 24.1
Печатается по рекомендации УМЦ КГТУ им. А.Н. Туполева, ИППК КГТУ им. А.Н. Туполева по направлению «Химия и инженерная экология», Научного Совета по неорганической химии (секция «Координационная химия») РАН
Рецензенты: проф. Г.К. Будников (Казанский государственный университет), проф. М.И. Евгеньев (Казанский государственный технологический университет)
Под редакцией проф. А.Н. Глебова
Авторский коллектив: А.Р. Буданов, Н.М. Воеводина, А.Н. Глебов,
И.Г. Григорьева, А.А. Кулаков, О.В. Лавриненко, С.А. Мальцева, И.П. Оранская,
А.И. Шамкаева, А.Г. Мельникова, А.В. Желовицкая, Э.В. Гоголь, Н.В. Кремлева, Е.Н. Офицеров, Д.В. Фролов, С.М. Шавалеева
Курс лекций по общей химии: Учеб. пособие для вузов. – 3-е изд., испр. и доп. – Казань: Изд-во «Экоцентр», 2005. – 136 с.
Учебное пособие предназначено для студентов технического университета, изучающих общую химию по дневной, вечерней и заочной формам обучения. Пособие составлено в соответствии с учебным планом, программой курса химии. «Курс лекций по общей химии» способствует более полному освоению изучаемого материала и развитию навыков самостоятельной работы студентов. В пособии приведены лекции по основным темам курса общей химии.
Лицензия Минпечати РТ
№ 0307 от 08.06.2000
Без объявл. 2005 © Издательство «Экоцентр», 2005.
Оглавление
| Предисловие | |
| Лекция 1. Предмет и значение химии | |
| Лекция 2. Основные законы химии | |
| Лекция 3. Строение атома | |
| Лекция 4, 5. Химическая связь | |
| Лекция 6. Периодический закон Д.И. Менделеева | |
| Лекция 7, 8. Химическая термодинамика | |
| Лекция 9. Химическая кинетика и катализ. Химическое равновесие | |
| Лекция 10. Растворы | |
| Лекция 11, 12. Растворы электролитов. Электролитическая диссоциация | |
| Лекция 13. Окислительно-восстановительные реакции | |
| Лекция 14. Электрохимические процессы и системы | |
| Лекция 15. Коррозия металлов и защита от коррозии | |
| Лекция 16. Координационные соединения | |
| Лекция 17. Обзор химических свойств элементов | |
| Дополнительные лекции | |
| К лекции 12. О структуре воды и растворов | |
| Лекция 18. Основы кристаллохимии | |
| Приложение | |
| Литература |
Предисловие
В условиях современной информатизации знаний представляется целесообразным в учебном процессе иметь краткие по объему, но значительные по содержанию учебники, позволяющие студентам более оперативно усваивать большой объем материала в соответствии с госстандартами и учебными программами. Особое значение попытка создания такого учебника приобретает в условиях сокращения числа аудиторных часов и одновременного увеличения часов, отведенных для самостоятельной работы, а также введения дистанционного обучения.
Учебное пособие достаточно полно отражает основной материал программы, характеризуется наглядностью, логичностью и последовательностью изложения.
Курс лекций был апробирован в течении длительного периода на кафедре общей химии и экологии КГТУ им А.Н. Туполева.
Авторский коллектив выражает признательность рецензентам и сотрудникам кафедры за помощь в создании книги.
Лекция 1
ПРЕДМЕТ И ЗНАЧЕНИЕ ХИМИИ
Современное определение химии: система химических наук (органическая, неорганическая, аналитическая, физическая химия и т.д.), главной задачей… Химия изучает состав, структуру веществ органического и неорганического… Значение химии в существовании и развитии человечества огромно и значимо. Достаточно сказать, что ни одна отрасль…Лекция 2
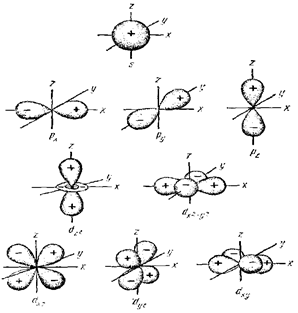
§ 1. Атомно-молекулярное учение – теоретический фундамент химии.Вещество – есть одна из форм существования материи. Вещество состоит из отдельных… Молекула – наименьшая частица вещества, обладающая его химическими свойствами.… Необходимо различать понятия простого (элементарного) вещества и химического элемента. В самом деле, каждое простое…Таблица 3.1
Электронные конфигурации атомов
Электронная конфигурация атома записывается в виде формулы, где количество электронов на подуровне указывается верхним индексом. Например, для… В многоэлектронном невозбужденном атоме электроны занимают орбитали с… Необходимо отметить, что для элементов с полностью или наполовину заполненными d- и f-подуровнями наблюдаются…Свойства элементов, простых тел и их соединений находятся в периодической зависимости от их атомных весов (масс).
Если в Периодической системе следующий по возрастанию атомного веса элемент не подходил по химическим свойствам, Менделеев оставлял в этом месте… Успех сформулированного Менделеевым периодического закона и таблицы элементов… § 2. Физическое обоснование Периодического закона. Открытие Д.И. Менделеева – пример гениального эмпирического…Классификация термодинамических систем
| Вид системы | Характеристика | Примеры |
| Открытая | Возможен обмен с внешней средой веществом и энергией | Человеческое тело |
| Замкнутая | Невозможен обмен с внешней средой веществом, возможен обмен энергией | Закрытая колба с реагентами |
| Изолированная | Невозможен обмен с внешней средой ни веществом, ни энергией | Термос, сосуд Дьюара |
| Адиабатически изолированная | Невозможен обмен с внешней средой тепловой энергией, возможен обмен веществом |
Одна и та же система может находиться в различных состояниях. Каждое состояние системы характеризуется определенным набором значений термодинамических параметров. К термодинамическим параметрам относятся температура, давление, плотность, концентрация и т.п. Изменение хотя бы только одного термодинамического параметра приводит к изменению состояния системы в целом. При постоянстве термодинамических параметров во всех точках системы (объема) термодинамическое состояние системы называют равновесным.
Различают гомогенные и гетерогенные системы. Гомогенные системы состоят из одной фазы, гетерогенные – из двух или нескольких фаз. Фаза – это часть системы, однородная во всех точках по составу и свойствам и отделенная от других частей системы поверхностью раздела. Примером гомогенной системы может служить водный раствор. Но если раствор насыщен и на дне сосуда есть кристаллы солей, то рассматриваемая система – гетерогенна (есть граница раздела фаз). Другим примером гомогенной системы может служить простая вода, но вода с плавающим в ней льдом – система гетерогенная.
§ 2. Внутренняя энергия. Первое начало (первый закон) термодинамики.Мерой внутренней энергии хаотического теплового (Броун) движения частиц в теле служит температура. Если тело А, вступая в контакт с телом В, отдает ему теплоту, то тело А имеет более высокую температуру, чем тело В. В тоже время нулевое начало термодинамики утверждает, что если тело А находится в тепловом равновесии (имеет одинаковую температуру) с телом В и телом С, то температура тел В и С также одинакова. Это начало лежит в основе измерения температуры при помощи термометра. При тепловом равновесии дальнейший обмен тепловой энергией невозможен.
Внутренняя энергия системы представляет собой ее полную энергию, которая складывается из кинетической и потенциальной энергий молекул, атомов, атомных ядер и электронов и представляет собой энергию поступательного, вращательного и колебательного движений частиц. Она не включает потенциальную энергию положения системы в пространстве и кинетическую энергию движения системы как целого. Иными словами потенциальная энергия (DU) вещества реализуется за счет разрыва и образования химической связи в теплоту (Q) и совершаемую работу (A).
Таблица 7.2
Классификация термодинамических процессов
| Процесс | Условия |
| Изотермический | Постоянная температура – T = const |
| Изобарный | Постоянное давление – p = const |
| Изохорный | Постоянный объем – V = const |
| Адиабатический | Отсутствие теплообмена между системой и внешней средой – dQ=0 |
Первое начало термодинамики, или закон сохранения энергии, гласит, что энергия не может возникать из ничего и исчезать, а только переходит из одной формы в другую. Например, внутренняя энергия, содержащаяся в веществе, может превращаться в тепловую, световую (пламя), электрическую (химический аккумулятор) и т.д.
Например, сообщим системе некоторое количество тепловой энергии Q, которая расходуется на совершение работы A и на изменение состояния внутренней энергии системы DU:
Q = A + DU (7.1),
где DU – изменение величины, которую называют внутренней энергией системы. Если система совершает работу за счет внутренней энергии без поступления теплоты, то A = –DU (адиабатический процесс).
Таким образом, увеличению внутренней энергии системы соответствуют положительные значения DU, уменьшению – отрицательные значения. Теплота, сообщаемая системе, и работа, совершаемая системой, также считаются
положительными, и наоборот, теплота, отдаваемая системой, и работа, совершаемая над системой, считаются отрицательными.
Внутренняя энергия относится к функциям состояния, т.е. величинам, которые зависят только от состояния системы и не зависят от ее предыстории. Следовательно, изменение внутренней энергии в ходе процесса зависит только от начального и конечного состояния и не зависит от пути протекания процесса. Напротив, работа зависит от пути протекания процесса, и поэтому не обладает свойствами функции состояния, как «запас работы», которую может совершить система.
Единицей измерения работы, теплоты и внутренней энергии в системе СИ служит джоуль (Дж). 1 джоуль – это работа силы в 1 ньютон на расстоянии 1 м (1 Дж = 1 Н×м = 1 кг×м2/с2). В старой химической литературе широко использовалась единица количества теплоты и энергии калория (кал). 1 Калория – это такое количество теплоты, которое необходимо для нагревания 1 г воды на 1°C. 1 Кал = 4,184 Дж » 4,2 Дж. Теплоты химических реакций удобнее выражать в кило-джоулях или килокалориях: 1 кДж = 1000 Дж, 1 ккал = 1000 кал.
§ 3. Закон Гесса. В химической термодинамике первый закон трансформируется в закон Гесса,характеризующий тепловые эффекты химических реакций.Теплота, как и работа, не является функцией состояния. Поэтому для придания тепловому эффекту свойства функции состояния введена энтальпия (DH), направленное изменение которой составляет DH = DU+PDV при постоянном давлении. Отметим при этом, что PDV = A – работе расширения, а DH = –Q (с обратным знаком). Энтальпию характеризуют теплосодержанием системы так, что экзотермическая реакция понижает DH. Обратите внимание, что выделению теплоты в химической реакции (экзотермической) соответствует DH < 0, а поглощению (эндотермической) DH > 0. В старой химической литературе была принята противоположная система знаков (!) (Q > 0 для экзотермических реакций и Q < 0 для эндотермических).
Изменение энтальпии (тепловой эффект) не зависит от пути реакции, а определяется только свойствами реагентов и продуктов (закон Гесса, 1836)
Покажем это на следующем примере:
C(графит) + O2(г.) = CO2(г.) DH1 = –393,5 кДж
С(графит) + 1/2 O2(г.) = CO(г.) DH2 = –110,5 кДж
СО(г.) + 1/2 O2(г.) = СО2(г.) DH3 = –283,0 кДж
Здесь энтальпия образования CO2 не зависит от того, протекает ли реакция в одну стадию или в две, с промежуточным образованием CO (DH1 = DH2 + DH3). Или иными словами сумма энтальпий химических реакций в цикле равна нулю:
rot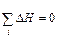 (7.2),
(7.2),
где i – число реакций в замкнутом цикле.
В любом процессе, когда конечное и начальное состояния веществ одинаковы, сумма всех теплот реакции равна нулю.
Например, мы имеем последовательность из нескольких химических процессов, приводящих в конце к исходному веществу и характеризующихся каждый своей энтальпией, т.е.
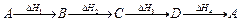 (7.3)
(7.3)
то, согласно закону Гесса,
DH1 + DH2 + DH3 + DH4 = 0 (7.4)
Результирующий тепловой эффект равен нулю потому, что на одних стадиях тепло выделяется, на других – поглощается, что приводит к взаимной компенсации.
Закон Гесса позволяет вычислить тепловые эффекты тех реакций, для которых прямое измерение невозможно. Например, рассмотрим реакцию:
H2(г.) + O2(г.) = H2O2(ж.) DH1 = ?
Экспериментально легко измерить следующие тепловые эффекты:
H2(г.) + 1/2O2(г.) = H2O(ж.) DH2 = –285,8 кДж
H2O2(ж.) = H2O(ж.) + 1/2O2(г.) DH3 = –98,2 кДж
Пользуясь этими значениями, можно получить:
DH1 = DH2 – DH3 = –285,8 + 98,2 = –187,6 (кДж/моль).
Таким образом, достаточно измерить тепловые эффекты ограниченного числа реакций, чтобы затем теоретически вычислить тепловой эффект любой реакции. На практике табулированы стандартные энтальпии образования DHf°298, измеренные при Т=298,15 К (25°C) и давлении p = 101,325 кПа (1 атм), т.е. при стандартных условиях. (Не путать стандартные условия с нормальными!)
Стандартная энтальпия образования DHf° – это изменение энтальпии в ходе реакции образования 1 моля вещества из простых веществ:
Ca (тв.) + C (графит) + 3/2O2 (г.) = CaCO3 (тв.) DH°298=–1207 кДж/моль.
Обратите внимание, что в термохимическом уравнении указываются агрегатные состояния веществ. Это очень важно, т.к. переходы между агрегатными состояниями (фазовые переходы) сопровождаются выделением или поглощением тепла:
H2 (г.) + 1/2O2 (г.) = H2O (ж.) DH°298 = –285,8 кДж/моль,
H2 (г.) + 1/2O2 (г.) = H2O (г.) DH°298 = –241,8 кДж/моль.
H2O (г.) = H2O (ж.) DH°298 = –44,0 кДж/моль.
Стандартные энтальпии образования простых веществ приняты равными нулю. Если простое вещество может существовать в виде нескольких аллотропных модификаций, то DH° = 0 приписывается самой устойчивой при стандартных условиях форме, например, кислороду, а не озону, графиту, а не алмазу:
3/2O2 (г.) = O3 (г.) DH°298 = 142 кДж/моль,
C (графит) = C (алмаз) DH°298 = 1,90 кДж/моль.
Следствием закона Гесса с учетом изложенного тогда изменение энтальпии в ходе реакции, будет равно сумме энтальпий образования продуктов за вычетом суммы энтальпий образования реагентов с учетом стехиометрических коэффициентов реакции:
DH(реакции) = SDHf(продуктов) – SDHf(реагентов) (7.5)
Вычислим стандартную энтальпию реакции:
4Al (тв.) + 3PbO2 (тв.) = 2Al2O3 (тв.) + 3Pb (тв.) (a)
Стандартные энтальпии образования найдем по справочным данным:
2Al (тв.) + 3/2O2 (г.) = Al2O3(тв.) DH°298 = –1669,8 кДж/моль, (b)
Pb (тв.) + O2 (г.) = PbO2 (тв.) DH°298 = –276,6 кДж/моль. (c)
С термохимическими уравнениями можно обращаться так же, как и с алгебраическими: умножать на число, складывать, вычитать, переносить слагаемые из одной части в другую.
4Al (тв.) + 3O2 (г.) + 3Pb (тв.) + 3O2 (г.) = 2Al2O3 (тв.) + 3PbO2 (тв.)
DH°298(a) = 2×DH°298(b) – 3×DH°298(c)
4Al (тв.) + 3PbO2 (тв.) = 2Al2O3 (тв.) + 3Pb (тв.)
DH°298(a) = 2×(–1669,8)–3×(–276,6) = –2509,8 кДж.
Обратите внимание, что свободный кислород, не участвующий в реакции, сократился при преобразованиях. На самом деле нет необходимости записывать расчеты так подробно. Если стехиометрические коэффициенты подобраны правильно, то баланс по всем веществам обязательно сойдется. Более кратко расчет DH реакции записывают так:
4Al(тв.) + 3PbO2(тв.) = 2Al2O3(тв.) + 3Pb(тв.)
ΔHf°298 0 3×(–276,6) 2×(–1669,8) 0
DH°298 = 2×(–1669,8)–3×(–276,6) = –2509,8 кДж.
Цикл Борна – Габера. Важным развитием и следствием закона Гесса является установление взаимосвязи между термодинамическими параметрами реакции, химических соединений и энергетическими параметрами атома, молекулы (потенциал ионизации, энергия сродства к электрону, энергия хим. связи, кристаллической решетки и др.). Так, в «Практикуме по общей химии» и в других учебниках дан пример взаимосвязи потенциала ионизации атома натрия (INa), энергий диссоциации молекулы хлора (DHдисс), сублимации (атомизации) натрия (DHат), сродства к электрону (ECl), кристаллической решетки хлорида натрия (U) и собственно энтальпии образования хлорида натрия (DHобр). Так что можно
записать
DHобр = DHат + INa + 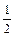 DHдисс + ECl + U (7.6)
DHдисс + ECl + U (7.6)
Ну, а вам останется только построить его графически еще раз.
Рассмотрим возможности цикла Борна – Габера на примере хлоридов (MeCl) и дихлоридов (MeCl2) металлов. Величина U рассчитывается на основе закона Гесса (с использованием старых единиц – ккал).
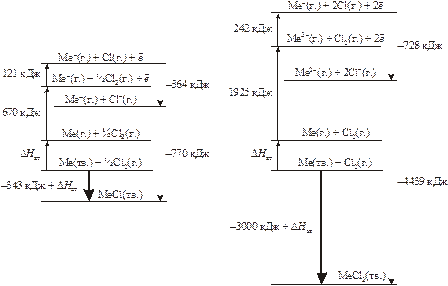
Какие же из хлоридов металлов более термодинамически устойчивы? В обоих случаях общей стадией является атомизация твердого металла с затратой
неизвестного нам, но одинакового количества энергии DHат. Очевидно, не эта величина определяет стабильность обоих хлоридов. Ионизация до двухвалентного состояния требует значительно большего количества энергии, чем однократная ионизация атома. На стадии ионизации хлора в случае MeCl2 также требуется затратить вдвое больше энергии, чем для MeCl. Энергетически выгодна форма MeCl2, а не MeCl. Хотя реакции 1-4 для MeCl и дают выигрыш
242 ккал/моль по сравнению с теми же реакциями для MeCl2, это преимущество полностью теряется на стадии образования кристаллической решетки из ионов. Выигрыш за счет энергии решетки MeCl2 оказывается 877 ккал/моль, что с избытком покрывает энергетические расходы на первых стадиях цикла. Поэтому, если даже соединение MeCl в какой-то момент возникает, то затем самопроизвольно по реакции диспропорционирования
2MeCl (тв.) = MeCl2 (тв.) + Me (тв.)
переходит в дихлорид. Тепловой эффект такого процесса будет равен
DH = –553 – DHат ккал/моль, т.е. очень большой величине, как раз и определяющей самопроизвольное течение реакции. Таким образом, можно без преувеличения сказать, что величина энергии кристаллической решетки определяет химическую или валентную форму ионного соединения.
Энергия кристаллической решетки в рядах однотипных соединений определяет также ряд свойств физико-химического характера, например температуру плавления и твердость.
Таблица 6.3.
Некоторые свойства галогенидов натрия.
| Энергия решетки, ккал/моль | Температура плавления, °С | Твердость, ед. Мооса * | |
| NaF | 3,25 | ||
| NaCl | 2,5 | ||
| NaBr | 2,25 | ||
| NaI | 2,0 |
* За 10 принимается твердость алмаза, за 1 – твердость талька.
Видно, что U и свойства находится в прямой зависимости от радиуса иона. Вам остается графически построить оба цикла и по справочным данным оценить энергетические параметры для хлоридов магния, кальция, бария.
§ 4. Второе начало (второй закон) термодинамики. Энтропия.Выделение тепловой энергии в ходе реакции способствует тому, чтобы она протекала самопроизвольно, т.е. без постороннего вмешательства. Однако имеются и другие самопроизвольные процессы, при которых теплота равна нулю (например, расширение газа в пустоту) или даже поглощается (например, растворение нитрата аммония в воде). Это означает, что помимо энергетического фактора на возможность протекания процессов влияет какой-то другой фактор.
Он называется энтропийным фактором или изменением энтропии. Энтропия S является функцией состояния и определяется степенью беспорядка в системе. Опыт, в том числе повседневный, свидетельствует о том, что беспорядок возникает самопроизвольно, а чтобы привести что-нибудь в упорядоченное состояние, нужно затратить энергию. Это утверждение – одна из формулировок второго начала термодинамики.
Существуют и другие формулировки, например, такая: Теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому (Клаузиус, 1850). Брусок, нагретый с одного конца, со временем принимает одинаковую температуру по всей длине. Однако никогда не наблюдается обратный процесс – равномерно нагретый брусок самопроизвольно не становится более теплым с одного конца и более холодным с другого. Другими словами, процесс теплопроводности необратим. Чтобы отнять тепло у более холодного тела, нужно затратить энергию, например, бытовой холодильник расходует для этого электрическую энергию.
Рассмотрим сосуд, разделенный перегородкой на две части, заполненные различными газами. Если убрать перегородку, то газы перемешаются и никогда не разделятся самопроизвольно снова. Добавим каплю чернил в сосуд с водой. Чернила распределятся по всему объему воды и никогда не соберутся
самопроизвольно в одну каплю. В обоих случаях самопроизвольно протекающие процессы сопровождаются увеличением беспорядка, т.е. возрастанием энтропии (DS > 0). Если мы рассматриваем изолированную систему, внутренняя энергия которой измениться не может, то самопроизвольность процесса в ней определяется только изменением энтропии: В изолированной системе самопроизвольно идут только процессы, сопровождающиеся возрастанием энтропии (Больцман, 1896). Это также одна из формулировок второго начала термодинамики. Наглядно проявление энтропийного фактора можно увидеть в фазовых переходах лед–вода, вода–пар, протекающие при постоянной температуре. Как известно при этом происходит поглощение (ледоход – похолодание) и выделение тепла (ледостав – потепление) так, что
DHГ = DS×T (7.7),
где DHГ – «скрытая» теплота фазового перехода. В фазовых переходах лед–вода–пар – DSл < DSв < DSп, т.е. энтропия возрастает при переходе от твердого тела к жидкости и от жидкости к газу, а ее величина тем больше, чем беспорядочнее движутся молекулы. Таким образом, энтропия отражает структурные отличия одного и того же химического элемента, молекулы, вещества. Например, для той же воды H2O – кристалл, жидкость, пар; для углерода – графит, алмаз, как аллотропные модификации и т.д.
Абсолютное значение энтропии можно оценить с использованием третьего начала термодинамики (постулата Планка), которое утверждает, что энтропия идеального кристалла при 0 К равна нулю lim DS=0 (при T®0).
Единицей измерения энтропии в системе СИ является Дж/К×моль. При этом понятно, что абсолютный нуль температуры недостижим (следствие из второго закона термодинамики), но он имеет важное значение в определении температуры – шкалы Кельвина.
Кроме того, есть еще одна функция состояния вещества – теплоемкость
DС = DH/DT, (7.8)
которая имеет такую же размерность, что и энтропия, но означает способность того или иного вещества отдавать (принимать) тепло при изменении температуры. Например, при изменении температуры на 100°С сталь быстрее нагреется и остывает соответственно, чем кирпич, поэтому печки кладут из кирпича. Величины теплоемкости удельные или молярные табулированы в справочниках обычно для изобарных условий – cp.
Стандартные абсолютные энтропии S°298 образования некоторых веществ приведены в справочной литературе. Обратите внимание, что в отличии от DHf простые вещества имеют значения S°298 > 0, т.к. их атомы и молекулы также находятся в беспорядочном тепловом движении. Чтобы найти изменение энтропии в реакции, можно также применить следствие закона Гесса:
DS(реакции) = SS(продуктов) – SS(реагентов) (7.9)
DS > 0 согласно второму началу термодинамики благоприятствует протеканию реакции, DS < 0 — препятствует.
Качественно можно оценить знак DS реакции по агрегатным состояниям реагентов и продуктов. DS > 0 для плавления твердых тел и испарения жидкостей, растворения кристаллов, расширения газов, химических реакций, приводящих к увеличению числа молекул, особенно молекул в газообразном состоянии. DS < 0 для сжатия и конденсации газов, затвердевания жидкостей, реакций, сопровождающихся уменьшением числа молекул.
Пользуясь справочными данными , рассчитаем DS°298 реакции (а).
4Al (тв.) + 3PbO2 (тв.) = 2Al2O3 (тв.) + 3Pb (тв.)
S°298 4×28,32 3×76,6 2×50,99 3×64,89
DS°298 = 2×50,99+3×64,89–4×28,32–3×76,6 = –46,4 Дж/К.
Таким образом, энтропийный фактор препятствует протеканию этой реакции, а энергетический фактор (см. выше) – благоприятствует.
Идет ли реакция на самом деле? Чтобы ответить на этот вопрос, нужно одновременно рассмотреть оба фактора: энтальпийный и энтропийный.
§ 5. Свободная энергия Гиббса. Критерии самопроизвольности протекания химических реакций.Одновременный учет энергетического и энтропийного факторов приводит к понятию еще одной полной функции состояния – свободной энергии. Если измерения проводятся при постоянном давлении, то величина называется свободной энергией Гиббса (в старой химической литературе – изобарно-изотермическим потенциалом) и обозначается DG.
Свободная энергия Гиббса связана с энтальпией и энтропией соотношением:
DG = DH – TDS (7.10)
где T – температура в кельвинах. Изменение свободной энергии Гиббса в ходе реакции образования 1 моля вещества из простых веществ в стандартных состояниях называется свободной энергией образования ΔG° и обычно выражается в кДж/моль. Свободные энергии образования простых веществ приняты равными нулю. Чтобы найти изменение свободной энергии Гиббса в ходе реакции, нужно от суммы свободных энергий образования продуктов отнять сумму свободных энергий образования реагентов с учетом стехиометрических коэффициентов:
DG(реакции) = SDG(продуктов) – SDG(реагентов) (7.11)
Самопроизвольным реакциям соответствует DG < 0. Если DG > 0, то реакция при данных условиях невозможна. Рассмотрим реакцию (a)
4Al (тв.) + 3PbO2 (тв.) = 2Al2O3 (тв.) + 3Pb (тв.)
ΔG°298 0 3×(–219,0) 2×(–1576,5) 0
ΔG°298 = 2×(–1576,5)–3×(–219,0) = –2496 кДж.
Существует и другой способ расчета DG реакции. Выше мы нашли значения ΔH и ΔS, тогда DG = DH – TDS
DG°298 = –2509,8 кДж – 298,15 К×(–0,0464 кДж/К) = –2496 кДж.
Таким образом, реакция (1) при стандартных условиях протекает самопроизвольно. Знак ΔG показывает возможность осуществления реакции только в условиях, для которых проводились вычисления. Для более глубокого анализа необходимо раздельное рассмотрение энергетического и энтропийного факторов. Имеется четыре возможных случая:
Таблица 7.3.
Определение возможности протекания химической реакции
| Критерии | Возможность |
| DH < 0, DS > 0 | Оба фактора благоприятствуют реакции. Как правило, такие реакции протекают быстро и полностью. |
| DH < 0, DS < 0 | Энергетический фактор благоприятствует реакции, энтропийный препятствует. Реакция возможна при низких температурах. |
| DH > 0, DS > 0 | Энергетический фактор препятствует реакции, энтропийный благоприятствует. Реакция возможна при высоких температурах. |
| DH > 0, DS < 0 | Оба фактора препятствуют реакции. Такая реакция невозможна. |
Если при стандартных условиях DG реакции > 0, но энергетический и энтропийный факторы направлены противоположно, то можно рассчитать, при каких условиях реакция станет возможной. DH и DS химической реакции сами по себе слабо зависят от температуры, если какие-нибудь из реагентов или продуктов не испытывают фазовых переходов. Однако в энтропийный фактор помимо DS входит также и абсолютная температура T. Таким образом, с повышением температуры роль энтропийного фактора повышается, и при температуре выше T » DH/DS реакция начинает идти в обратном направлении.
Если DG = 0, то система находится в состоянии термодинамического равновесия, т.е. DG – термодинамический критерий химического равновесия реакций (смотри вышеприведенные фазовые переходы воды).
Итак, анализируя функции состояния системы – энтальпию, энтропию и свободную энергию Гиббса – и их изменение в ходе химической реакции, можно определить, будет ли данная реакция происходить самопроизвольно, и тем самым ответить на вопросы, поставленные в начале этой лекции.
Более полно термодинамика отражена в курсе физической химии, а с прикладными ее аспектами вы ознакомитесь в курсах теплотехники и др.
Лекция 9
Химическая кинетика И катализ.
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Как протекают реакции во времени? Закономерностями протекания процессов во времени занимается химическая кинетика. Для управления химическими процессами необходимо знание скоростей химических реакций и факторов, влияющих на скорость.
§ 1. Скорость химической реакции. Основным понятием химической кинетики является скорость реакции. Скоростью химической реакции называют изменение количества реагирующего вещества или образующегося продукта в единице объема за единицу времени.
Для гомогенной (однофазной) системы реакционным пространством служит объем, и скорость v может быть выражена:
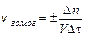 ;
; 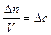 ;
; 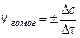 ;
; 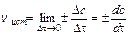 (9.1)
(9.1)
где Dn – изменение количества вещества; t – время; Dc – изменение концентрации. В кинетике под реагентами понимают исходные вещества, а образующиеся в ходе реакции вещества – продукты.
Изменение концентрации веществ показаны на рис. 9.1. Видно, что u для исходных веществ имеет знак (–) минус.
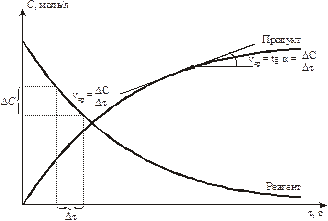
Рис. 9.1. Изменение концентраций реагентов и продуктов при химическом взаимодействии.
Для гетерогенных систем (различные фазы) реакции протекают на поверхности раздела фаз, и скорость определяется количеством вещества, вступившего
в реакцию или получившегося в результате реакции (Dn) за единицу времени (Dt) на единице поверхности раздела (S):
 ;
; 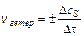 ;
; 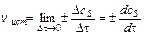 (9.2)
(9.2)
Основной закон химической кинетики – закон действующих масс (ЗДМ), высказанный в 1865 г. М.Н. Бекетовым и уточненный норвежскими химиками Гульдбергом и Вааге. Зависимость скорости химической реакции от концентрации выражается законом действующих масс: скорость простой химической реакции прямо пропорциональна концентрации реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции (aA+bB=dD), или в математическом виде:
 (9.3)
(9.3)
где k – константа скорости химической реакции, равная скорости химической реакции при концентрациях реагирующих веществ 1 моль/л.; a и b – частные порядки реакции, С – концентрации.
Например, для реакции 2А+В=А2В скорость определится следующим
образом:
v = k CA2 CB (9.4)
Константа скорости не зависит от концентрации веществ, а зависит от температуры. Выше указанное отношение (9.3) справедливо только для гомогенных реакций. Для гетерогенных процессов концентрация вещества в твердой фазе не меняется, поэтому скорость определяется концентрацией газов или растворенных веществ. Так, для реакции С+О2=СО2
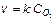 (9.5)
(9.5)
Обычно концентрацию газов заменяют парциальным давлением газа PO2. Численное значение k при этом станет другим.
Классификация химических реакций. В химической кинетике реакции классифицируют по I. молекулярности и порядку реакции; II. по механизму протекания.
I. 1) Молекулярность определяется числом молекул, участвующим в элементарном химическом акте:
Н2 = 2Н – мономолекулярная;
CН3COOC2H5 + H2O = CН3COOH + C2Н5OH – бимолекулярная;
2NO + Cl2 = 2NOCl – тримолекулярная.
Реакции с молекулярностью большей трех не встречаются, т.к. вероятность столкновения сразу четырех и более молекул очень мала.
2) Порядок реакции – это сумма показателей степеней при концентрациях веществ в уравнении закона действующих масс. Так, реакции (9.5) – первого порядка, реакция (9.4) – третьего порядка. Иногда указывают частный порядок реакции по веществу – это степень, в которую возводится концентрация этого вещества.
II. 1) Параллельные реакции. Практически выгодным является получение перхлората калия, который является одним из компонентов ракетного топлива, поэтому изучение скорости и факторов влияющих на скорость химических реакций позволяет управлять параллельными реакциями и достигнуть v1 < v2.
2KOH + Cl2  KClO + KCl, 6KOH + Cl2
KClO + KCl, 6KOH + Cl2  KClO + 5KCl
KClO + 5KCl
2) Последовательные реакции А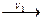 В
В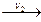 С
С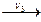 D. Чтобы управлять процессом, необходимо выявить стадию, у которой скорость реакции минимальна, то есть скорость реакции, которая является лимитирующей для всего процесса. Затем, подбирая условия реакции, увеличивать скорость лимитирующей стадии, тем самым будет увеличиваться скорость всего процесса.
D. Чтобы управлять процессом, необходимо выявить стадию, у которой скорость реакции минимальна, то есть скорость реакции, которая является лимитирующей для всего процесса. Затем, подбирая условия реакции, увеличивать скорость лимитирующей стадии, тем самым будет увеличиваться скорость всего процесса.
3) Сопряженные реакции. Одна реакция идет только в том случае, если идет вторая.
§ 2. Уравнение Аррениуса. Скорость химической реакции зависит от температуры. При повышении температуры скорость большинства химической реакций увеличивается. Вант-Гофф показал, что при повышении температуры на 10 град., скорость реакции увеличивается в 2-4 раза (правило Вант-Гоффа):
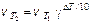 (9.6)
(9.6)
Температурный коэффициент реакции g равен отношению скоростей реакции, когда Т2–Т1=10 град. и показывает, во сколько раз увеличивается скорость реакции при повышении температуры на 10 град. Но уравнение (9.6) лишь приблизительно оценивает зависимость скорости реакции и константы скорости от температуры. Функциональную зависимость константы скорости реакции от температуры установил Аррениус (9.7).
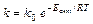 (9.7)
(9.7)
где k0 – начальная скорость или предэкспоненциальный множитель – А. Графически величину Еа по Аррениусу можно представить на рис. 9.2.
В логарифмической форме это уравнение для констант скоростей преобразуется:
ln k = –Ea/RT + ln k0 (9.8)
Видно, что зависимость константы скорости реакции от температуры линейна, следовательно можно определить энергию активации по тангенсу угла наклона прямой и k0 – по отрезку, отсекаемому прямой по оси ординат, когда 1/Т→0. Энергию активации легко рассчитать, если известны константы скорости при разных температурах.
Все молекулы, запас энергии, которых не ниже энергетического барьера называются активными и они находятся в состоянии активированного комплекса. В состоянии активированного комплекса нет уже исходных веществ, но и еще нет продуктов реакции (старые связи еще не до конца разорвались, а новые
не образовались). Энергия такой системы максимальна, а значит, активированный комплекс неустойчив.
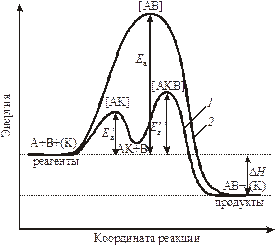
Рис. 9.2. Энергетический профиль реакции: 1 – в присутствии катализатора; 2 – без катализатора. Под координатой реакции понимается многопараметровая функция, зависящая от времени, пространственных координат молекул реагентов и др.
Разница в энергиях между исходным и переходным состоянием равна энергии активации. То есть, активные молекулы должны обладать энергией активации для осуществления взаимодействия для того, чтобы произошло ослабление связей в исходных веществах, для преодоления отталкивания между электронами в сближающихся молекулах, которое мешает их столкновению. Следовательно, энергия активации является одним из параметров, характеризующих скорость процесса. Чем больше энергия активации, тем меньше скорость процесса. Если Еа больше 1000 кДж/моль, то реакция не протекает при комнатной температуре, такие реакции идут при особых условиях, высоких Т и Р. Энергия активации в первую очередь зависит от природы реагирующих частиц. Существуют реакции, у которых Еакт = 0 – это туннельные реакции, протекающие, как правило, при низких температурах.
§ 3. Влияние катализаторов на скорость химической реакции. Катализаторами называются вещества, увеличивающие скорость реакции, но не расходующиеся в результате ее протекания. Катализаторы обычно участвуют в образовании промежуточных соединений, но в конце реакции полностью регенерируются. Наряду с катализаторами существуют ингибиторы, замедляющие скорость реакции. Катализ – наиболее эффективный метод интенсификации промышленных химических процессов. С точки зрения закона Аррениуса катализаторы (К) снижают энергетический барьер, а ингибиторы увеличивают (рис. 9.2).
1) Катализ применим только тогда, когда свободная энергия Гиббса данной реакции отрицательна (DG<0).
2) В присутствии катализатора изменяется механизм химической реакции, она протекает через новые стадии, характеризующиеся небольшими значениями Еа.
3) При катализе не изменяется тепловой эффект реакции.
4) Если реакция не обратима, то катализатор не влияет на положение равновесия.
5) Катализаторы обычно действуют избирательно, селективно. Одним из универсальных катализаторов является вода.
Активность того или иного катализатора зависит от многих факторов. Активность катализатора может быть повышена специальными добавками – промоторами. Если катализатор и реагирующие вещества образуют одну фазу, то катализ называют гомогенным, если две фазы – гетерогенным. Для объяснения гомогенного катализа используют теорию промежуточных соединений. Согласно этой теории катализатор взаимодействует с одним из исходных веществ с образованием промежуточных соединений, при этом понижается энергия активации и растет скорость реакции. А+В=AB; А+К=[АК]; [АК]+В=AB+К.
Для своего прохождения реакция должна преодолеть «энергетический барьер» – энергию активации и чем меньше барьер, тем больше скорость реакции. Таким образом, скорость реакции определяется числом активных молекул и величиной энергии активации, последняя изменяется в пределах 40-350 кДж.
§ 4. Химическое равновесие. Химические реакции бывают необратимые (односторонние) и обратимые (двухсторонние) или равновесные. Необратимые реакции протекают только в одном направлении до полного превращения хотя бы одного из реагирующих веществ. Примером необратимой реакции может служить реакция разложения бертолетовой соли:
2KClO3 = 2KCl + 3O2
Обратимые реакции могут протекать как в прямом, так и в обратном направлении, в связи с этим ни одно из реагирующих веществ не расходуется полностью. Так, например, следуя рис. 9.1, в состоянии равновесия имеет место определенное количество как реагента, так и продукта. Например, реакция синтеза аммиака является обратимой:
3Н2 + N2 ⇄ 2NН3
Она может протекать как в сторону образования аммиака (прямой процесс), так и в сторону реагентов – молекулярного азота и водорода (обратный процесс). Знак обратимости реакции – стрелки с противоположными направлениями: ⇄.
Выразим для обратимого процесса две скорости прямой ( ) и обратной реакции (
) и обратной реакции (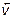 ) по закону действующих масс:
) по закону действующих масс:
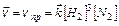 (9.9)
(9.9)
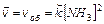 (9.10)
(9.10)
Когда скорости прямой и обратной реакций становятся равными, наступает химическое равновесие. Комбинируя уравнения 9.9, 9.10, получим:
 (9.11)
(9.11)
Это уравнение отвечает кинетическому условию равновесия. Химическое равновесие называют динамическим. Этим подчеркивается, что при равновесии протекают как прямая, так и обратная реакции, но их скорости равны, а устанавливается оно с течением времени.
Кравн – константа равновесия является количественной мерой, характеризующей реакцию в состоянии химического равновесия. Закон действующих масс для обратимого процесса можно сформулировать: В состоянии химического равновесия отношение произведения концентраций продуктов реакции к произведению концентраций исходных веществ есть величина постоянная. При этом концентрация каждого вещества берется в степени его стехиометрического коэффициента в уравнении реакции. Значения некоторых Кравн приведены в «Практикуме».
Система находится в состоянии равновесия до тех пор, пока внешние условия сохраняются постоянными. Если же условия изменятся, то система выйдет из равновесия. При этом скорости прямого и обратного процессов изменяются неодинаково, и будет протекать преимущественно только один процесс (прямой или обратный), т.е. произошло смещение равновесия. Оно происходит под действием таких факторов: изменение концентрации веществ, температуры, давления и подчиняется универсальному принципу Ле-Шателье: Всякое внешнее воздействие на равновесную химическую систему вызывает в ней процесс, приводящий к уменьшению эффекта внешнего воздействия, который часто называют «третьим законом Ньютона в химии» и имеющий универсальный характер в природе.
Влияние изменения концентрации веществ на смещение равновесия. По принципу Ле-Шателье увеличение концентрации исходных веществ сместит равновесие в сторону той реакции, при которой происходит уменьшение количества исходных веществ, т.е. в сторону превращения исходных веществ в продукт реакции.
Наоборот, увеличение концентрации продуктов реакции сместит равновесие в сторону образования исходных веществ. Уменьшение же концентрации продуктов реакции сместит равновесие в сторону образования продуктов реакции и концентрация их возрастет.
Обобщая, можно сделать вывод, что при увеличении концентрации какого-либо из веществ равновесие смещается в сторону расхода этого вещества;
при уменьшении концентрации какого-либо вещества равновесие смещается в сторону образования этого вещества.
Равновесие подавляющего большинства реакций смещается при изменении температуры. Фактором, определяющим направление смещения равновесия, является знак теплового эффекта реакции. Из принципа Ле-Шателье следует, что при повышении температуры равновесие смещается в сторону эндотермической реакции, а при охлаждении – в сторону экзотермической реакции.
В рассматриваемом случае синтез аммиака представляет собой экзотермический процесс: DH=–92 кДж/моль. Поэтому при повышении температуры равновесие смещается в сторону обратной реакции, разложения аммиака.
В соответствии с принципом Ле-Шателье при увеличении давления в системе путем сжатия равновесие сдвигается в сторону уменьшения числа молей газа; при уменьшении давления равновесие сдвигается в сторону возрастания числа молей газов.
Например, при синтезе аммиака при протекании прямого процесса из четырех молей газа образуется два моля газа, значит число молей газа уменьшается в два раза, что приводит к уменьшению давления в сосуде. При обратном процессе число молей газа наоборот возрастает, следовательно, возрастает и давление. Отсюда, вывод: синтез аммиака нужно вести при высоких давлениях. В том случае, когда реакция протекает без изменения числа молей газа, равновесие при изменении давления не нарушается. Напомним, что термодинамическим критерием химического равновесия является DG=0, что особенно наглядно проявляется при фазовых переходах.
Термодинамические и кинетические критерии химического равновесия связаны уравнением:
DG = –RT ln Kравн (9.12).
Из уравнения видно, что высоким отрицательным значениям DG отвечает большое значение константы равновесия (Kp), это значит, что в равновесной смеси преобладают продукты реакции. Если же DG имеет большое положительное значение (Kp), то в равновесной смеси преобладают исходные вещества. Уравнения (9.9) позволяют по величине DG вычислить константу равновесия (Кравн), а затем и равновесные концентрации или парциальные давления реагентов.
Таким образом, количественно химическое равновесие оценивается термодинамическим критерием (DG) и кинетическим критерием (равенство скоростей прямой и обратной реакций, Kp).
Лекция 10
Растворы
§ 1. Определение растворов. Растворимость. Способы выражения концентрации.Для того чтобы между двумя веществами произошла химическая реакция, их молекулы должны вступить в контакт друг с другом. Так, если измельчить твердые вещества в порошок, то реакции между ними ускорятся во много раз. Растворяя эти вещества, мы дробим их до наименьших возможных частиц – молекул или ионов, поэтому большинство химических реакций оказывается выгодным проводить в растворах. Реакции, протекающие в растворах, имеют важное значение для биохимии, химической технологии и т.д. Современное учение о растворах создавалось на протяжении примерно двухсот лет, в него внесли вклад такие знаменитые ученые, как М. Фарадей, Гельмгольц, Д.И. Менделеев, Аррениус, Вант-Гофф, Оствальд, Каблуков и другие. Современное определение понятия раствор следующее:
Раствором называется гомогенная многокомпонентная химическая система, состав которой в определенных пределах может варьироваться (быть переменным) без качественного изменения свойств.
Это определение требует некоторых комментариев. Гомогенная – значит, ее свойства однородны по всему объему, т.е. отсутствуют границы раздела фаз; многокомпонентная – означает совокупность нескольких веществ: растворителя и одного или нескольких растворенных веществ – компонентов. Химическая система переменного состава означает взаимодействие растворителя и компонентов с образованием химических соединений переменного состава.
Химическое взаимодействие растворителя с компонентами называется сольватацией, а в случае растворителя воды – гидратацией. Процесс этот сопровождается поглощением или выделением тепла, как и в других химических реакциях. Образующиеся сольваты и гидраты в растворе в зависимости от концентрации, температуры, давления и других факторов имеют переменный состав в отличии от исходных реагентов: растворителя и компонентов. Например, H2O и C2H5OH – соединения постоянного состава образуют сольваты {(H2O)x× (C2H5OH)y}, где x и y – целые числа (1,2,3) – соединения переменного состава. Растворы как бы занимают промежуточное положение между смесями и химическими соединениями. Растворы обладают определенной структурой, особенно для ближайшего окружения сольватов.
Важной количественной характеристикой растворов являются концентрация и растворимость. Под концентрацией понимается количество растворенного вещества в объеме раствора (растворителя). Под растворимостью понимается максимально возможное количество растворенного вещества в объеме (массе) растворителя до появления осадка (гетерогенная система, и есть граница раздела фаз). Естественно, они зависят от температуры и других внешних факторов. Растворы могут быть твердыми, жидкими и газообразными. Они также классифицируются на растворы неэлектролитов и электролитов, о которых будет идти речь в следующих лекциях.
Как правило, растворимость твердых веществ повышается с ростом температуры (хотя есть исключения) и мало зависит от изменения внешнего давления. Растворимость газов также варьирует в широких пределах (от 0,2 объема на
1 объем воды у гелия до 800 объемов у аммиака), однако понижается с ростом температуры и повышается с ростом парциального давления газа.
В случае ограниченной растворимости можно выделить растворы ненасыщенные, содержание растворенного вещества в которых меньше, чем максимально возможное при данных условиях; насыщенные, в которых данное вещество при данных условиях больше не растворяется, и пересыщенные, содержание растворенного вещества в которых больше, чем в насыщенном растворе при данных условиях (пересыщенный раствор можно получить, например, быстрым охлаждением раствора вещества, растворимость которого значительно повышается с ростом температуры). Пересыщенные растворы метастабильны, т.е. способны под действием внешних факторов, а иногда и самопроизвольно, переходить в устойчивое состояние (насыщенный раствор + избыток растворенного вещества в отдельной фазе).
Способы выражения концентрации растворов даны в «Практикуме». Значения растворимостей различных веществ в различных растворителях табулированы в многочисленных химических справочниках.
§ 2. Термодинамические свойства растворов.Для растворов характерны несколько законов. К ним относятся: понижение давления пара растворителя, повышение температуры кипения и понижение температуры замерзания раствора, осмотическое давление и другие, связывающие концентрацию, растворимость с физическими параметрами раствора, как химической системы.
Величина относительного понижения давления пара (депрессии) над раствором по сравнению с чистым растворителем пропорциональна концентрации растворенного вещества (закон Рауля):
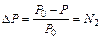 , (10.1)
, (10.1)
где P0 – давление пара над чистым растворителем, P — давление пара над раствором, N2 – мольная доля растворенного вещества.
Физическую природу этого явления можно пояснить следующим образом. В замкнутой системе, состоящей из жидкого растворителя и паров над ним, при постоянных условиях устанавливается динамическое равновесие между жидкой и газообразной фазой: количество молекул, перешедших из газообразной фазы в жидкую за данный промежуток времени, равно количеству молекул, перешедших из жидкой фазы в газообразную. Первая величина зависит от содержания молекул растворителя в единице объема газовой фазы, вторая — от количества молекул растворителя на единицу поверхности раздела фаз. Добавление
нелетучего растворенного вещества уменьшает только вторую величину (часть поверхности занята молекулами растворенного вещества), поэтому положение равновесия должно сместиться в сторону уменьшения давления пара растворителя. Т.е. давление пара воды над морской водой меньше чем над пресной. У закона Рауля есть два следствия, связанные с кипением и замерзанием (отверждением) растворов по сравнению с растворителем.
Повышение температуры кипения растворов, по сравнению с растворителем называется эбулиоскопией, а математически выражается формулой:
DTкип = KE×Cm, (10.2)
где KE – эбулиоскопическая постоянная для данного растворителя, Сm – моляльная концентрация. Аналогично для замерзания растворов – криоскопия и уравнения для температуры замерзания раствора по сравнению с чистым растворителем и имеет вид:
DTзам = KE×Cm, (10.3)
где КК – криоскопическая постоянная. Методами эбулиоскопии и криоскопии можно, например, определить молекулярную массу неизвестного вещества. Как известно, если к g1 г растворителя добавить g2 г растворенного вещества, то моляльная концентрация будет равна:
 , (10.4)
, (10.4)
где M – молекулярная масса растворенного вещества.
Тогда из уравнений (10.2)-(10.4) следует, что
 или
или 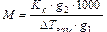 (105)
(105)
Наглядно следствия из закона Рауля объясняются изменениями на диаграмме состояния воды и раствора (рис. 10.1).
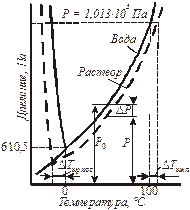
| Рис. 10.1. Фазовая диаграмма воды и раствора при постоянной концентрации. |
При пониженном по сравнению с чистым растворителем давлении паров над раствором «ветвь» граница раздела фаз понижается с увеличением концентрации растворенного вещества. При этом раствор кипит при большей температуре, чем растворитель, а замерзает при более низкой.
У эбулиоскопии и криоскопии очень много прикладных значений (посыпание солью снега зимой, охлаждающие смеси, высокотемпературные жидкости и т.д.).
У закона Рауля есть «родственник» – закон Генри: растворимость газов в жидкости пропорционально давлению:
C = k×p, (10.6)
где k – константа Генри, p – давление. Он наглядно иллюстрируется на примерах: сифон, газированная вода, наличием кислорода на больших водных глубинах, водолазная техника («кессонная болезнь») и т.д.
И наконец рассмотрим еще один закон – закон осмотического давления Вант-Гоффа.
Pp = C×R×T, (10.7)
где Pp – осмотическое давление, C – молярная концентрация, R – газовая постоянная, T– температура; напоминающий уравнение Клайперона – Менделеева, но связанный с явлением осмоса: давление растворителя над полупроницаемой мембраной выше, чем в растворе (с другой стороны мембраны). Не вдаваясь в суть явления осмоса, отметим, что под полупроницаемой понимается мембрана, проницаемая для молекул растворителя, но задерживающая молекулы растворенного вещества. Закон также имеет большое прикладное значение в природе и технике: рост растений, деревьев, очистка жидкостей и т.д.
Перечисленные термодинамические законы являются общими для всех растворов нелетучих веществ.
Лекция 11, 12
РАСТВОРЫ ЭЛЕКТРОЛИТОВ.
электролитическАЯ диссоциациЯ
Вещества, растворы (или расплавы) которых проводят электрический ток, называются электролитами, для которых Фарадей в начале XIX в. ввел понятие катион и анион и установил законы электролиза. Носителями электричества в растворе являются катионы и анионы, т.е. атомы и молекулы несущие положительный и отрицательный заряд, по Фарадею возникающие в процессе прохождения электрического тока. Лишь спустя почти полвека великий Сванте Аррениус установил, что все как раз наоборот: ионы (катионы, анионы) образуются в растворах электролитов всегда вследствие электролитической диссоциации полярных молекул и ионных кристаллов.
§ 1. Основные положения теории электролитической диссоциации.При растворении солей, кислот и оснований в воде происходит диссоциация этих веществ с образованием электрически заряженных частиц – катионов и анионов; электрическая проводимость водных растворов солей, кислот и оснований пропорциональна общей концентрации ионов в растворе. Таким образом, ионы самопроизвольно, изначально существуют в растворе электролитов, так что в целом раствор электронейтрален, а при наложении разности потенциалов проводит электрический ток. Аррениус ввел представление об электролитической диссоциации для объяснения природы изотоническогоё коэффициента – i, эмпирически в веденного Вант-Гоффом для выполнения термодинамических законов растворов электролитов, т.е. для законов Рауля, эбулиоскопии, криоскопии, Вант-Гоффа (10.1-10.5) он предложил ввести изотонический коэффициент – множитель концентрации. Так что, например, для осмотического давления следует писать
p = iCRT, (11.1)
С введением изотонического коэффициента эффективная концентрация электролита увеличивалась. По Аррениусу она увеличивалась за счет диссоциации – распада молекулы на несколько ионов.
Аррениус ввел понятие – степень электролитической диссоциации (a) как отношение числа молекул, продиссоциированных на ионы к общему числу молекул в растворе. Она меняется от 0 до 1, или от 0 до 100%.
Вспомните слабые, сильные электролиты, теперь они характеризуются a, которая связана с изотоническим коэффициентом следующим соотношением:
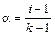 , (11.2)
, (11.2)
где i = 1 для растворов неэлектролитов, i > 1 для растворов электролитов. С уменьшением концентрации i увеличивается  ; k – целое число ионов в результате полной диссоциации, для бинарных электролитов (KCl, HCl) k = 2, для H3PO4: k=4, i<4 и т.д.
; k – целое число ионов в результате полной диссоциации, для бинарных электролитов (KCl, HCl) k = 2, для H3PO4: k=4, i<4 и т.д.
Таким образом, в растворах электролитов имеет место гидратация и сольватация (Менделеев) и электролитическая диссоциация (Аррениус) как два единых процесса растворения электролитов в растворе («две стороны одной медали»), что и ввел в теорию и практику Каблуков.
Электролитическая диссоциация вызывается взаимодействием полярных молекул растворителя с частицами растворяемого вещества. Это взаимодействие приводит к поляризации связей. Происходит образование ионов за счет «ослабления» и разрыва связей в молекулах растворяемого вещества. Переход ионов в раствор сопровождается их гидратацией:
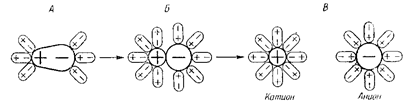
Рис. 11.1. Схема процессов гидратации и диссоциации полярной молекулы электролита типа HCl (разрыв ионогенной полярной ковалентной связи с ее переходом в ионную связь).
Кратко остановимся на водных растворах электролитов, основными участниками которых являются вода, кислоты, щелочи, соли и образуемые ими гидраты ионов (аквакомплексы), комплексные соединения.
Особенностью водных растворов электролитов является ион водорода (протон), имеющий малый размер (10–4 от размера атома), и образующий при гидратации ион гидроксония – Н3О+. Новая ковалентная связь образуется по донорно-акцепторному механизму за счет свободной электронной пары кислорода (донора) и вакантной орбитали протона (акцептора).
Характер ионов, образующихся при диссоциации различных электролитов, естественно, должен быть различным. В молекулах солей диссоциация всегда приводит к образованию катионов металла и анионов кислотного остатка. Кислотами являются электролиты, которые диссоциируют с образованием ионов Н+. Сильные кислоты (HCl, HBr, HI, HNO3, H2SO4) диссоциируют практически полностью, у слабых кислот диссоциирована лишь часть молекул. Основание определяют как электролит, диссоциирующий с образованием ионов ОН–. Сильные основания (NaOH, KOH) диссоциируют практически полностью, у слабых электролитов диссоциирована лишь некоторая доля молекул.
Существуют электролиты, которые могут диссоциировать как кислоты и как основания. Такие электролиты называются амфотерными электролитами (амфолитами). Амфотерность электролитов объясняется малым различием прочности связей Me–O и H–O. Примером амфотерного электролита может быть гидроксид цинка:
2H++ZnO22– ⇄ H++HZnO2– ⇄ Zn(OH)2 ⇄ZnOH++OH– ⇄Zn2++2OH–
При взаимодействии с азотной кислотой
Zn(OH)2 + 2HNO3 = Zn(NO3)2 + 2H2O
при взаимодействии с гидроксидом калия
Zn(OH)2 + 2KOH = K2ZnO2 + 2H2O
В растворах слабых электролитов процесс диссоциации протекает обратимо и, следовательно, к нему может быть применен закон действия масс, например:
CH3COOH ⇄ CH3COO–+ H+
Константа равновесия Kp будет равна:
 (11.3).
(11.3).
Константа равновесия для процесса диссоциации называется константой диссоциации Kдис.
Установлено, что многоосновные кислоты отщепляют ион водорода не сразу, а постепенно. Например,
1) H3PO4 ⇄ H++H2PO4– K1=7,11×10–3
2) H2PO4 ⇄ H++HPO42– K2=6,34×10–8
3) HPO4 ⇄ H++PO43– K3=1,26×10–12
Каждая ступень характеризуется своей константой диссоциации, но первая ступень идет легче, поэтому К1 больше следующей и т.д.
Константа диссоциации представляет собой важную характеристику слабых электролитов, так как указывает на прочность их молекул в данном растворе. Чем меньше константа диссоциации в данном растворителе, тем слабее диссоциирует электролит и тем устойчивее его молекулы.
Константа электролитической диссоциации зависит лишь от природы электролита и температуры. Между константой и степенью электролитической диссоциации существует количественная связь. Действительно, пусть в рассмотренном процессе общая концентрация растворенного вещества КА равна C, а степень диссоциации равна a. Тогда [K+] = [A–] = a×C и соответственно концентрация недиссоциированных частиц [KA] = (1–a)×C. Подставив эти значения в выражение для константы диссоциации, получим известный закон разбавления Оствальда:
 (11.4)
(11.4)
Это соотношение называется законом разбавления Оствальда. Для слабых электролитов, когда a <<1, Кдис ≈ a2C. Отсюда
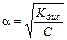 или
или 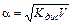 , (11.5)
, (11.5)
где V=1/C – разбавление. Из формулы следует, что при разбавлении раствора в 100 раз степень диссоциации возрастет в 10 раз.
Электропроводность растворов электролитов зависит от их концентраций в растворе. Величина, характеризующая данный раствор, называется удельной электропроводностью c
c = 1 / r (11.6)
где r – удельное электрическое сопротивление.
С разбавлением раствора она уменьшается, вследствие уменьшения концентрации, но в тоже время увеличивается степень диссоциации.
§ 2. Сильные электролиты. Активность. Согласно теории диссоциации, сильные электролиты практически полностью диссоциируют не только в разбавленных растворах, но и в растворах любой концентрации. Другими словами, в любых растворах сильных электролитов степень их диссоциации a =1 (100%).
Таким образом, считается, что в растворах сильных электролитов нет недиссоциированных молекул, поэтому понятие константы диссоциации к ним не применимо (знаменатель выражения вида (11.3) равен нулю).
При определенных условиях, например, когда растворитель обладает малой диэлектрической проницаемостью, создаются условия для электростатического взаимодействия сольватированных ионов противоположного знака. При этом последние подходят друг к другу на близкое расстояние и образуют так называемую ионную пару – сложный агрегат, состоящий из двух противоположно заряженных ионов, окруженных молекулами растворителя, в котором электрические заряды взаимно компенсированы.
При повышении концентрации раствора расстояния между ионами сокращаются, что усиливает межионное взаимодействие. Вследствие этого экспериментально определяемые свойства растворов сильных электролитов, зависящие от общего количества частиц в растворе, оказываются меньше рассчитанных в предположении полной диссоциации.
Чтобы можно было пользоваться простыми соотношениями идеальных растворов для описания поведения реальных растворов, Льюис (1907) ввел формальное представление об эффективной концентрации – активности. Активность связана с истинной концентрацией растворенного вещества соотношением
a = f×C, (11.7)
где a – активность, f – коэффициент активности, C – концентрация.
Активность выражается в тех же единицах, что и концентрация, поскольку коэффициент активности – величина безразмерная. Он характеризует степень отклонения свойств данного раствора от свойств идеального раствора. Для бесконечно разбавленных растворов электролитов, где практически отсутствует взаимодействие ионов, активность становится равной концентрации и коэффициент активности равен единице.
Так как коэффициенты активности учитывают также степень диссоциации электролита в растворе, их применяют и при описании свойств растворов слабых электролитов. Коэффициенты активности вычисляют по экспериментальным данным. Для этого измеряют какое-либо из свойств раствора (например, электропроводность, температуру кипения или замерзания) и определяют коэффициент активности как частное от деления экспериментально полученной величины на теоретически рассчитанную по законам идеальных растворов:
Таблица 11.1
Коэффициенты активности некоторых электролитов в растворах при 298 К
| Концентрация, | f ± для электролитов | ||||||
| NaCl | KCl | NaOH | KOH | HCl | H2SO4 | CaCl2 | |
| 0,001 0,01 0,1 0,5 1,0 2,0 5,0 | 0,965 0,874 0,778 0,681 0,657 0,668 0,874 | 0,966 0,901 0,769 0,651 0,607 0,576 — | 0,966 0,900 0,776 0,693 0,679 0,700 1,060 | 0,966 0,900 0,766 0,712 0,735 0,863 1,670 | 0,966 0,904 0,796 0,758 0,809 1,010 2,380 | 0,830 0,544 0,265 0,156 0,132 0,128 0,208 | 0,840 0,850 0,518 0,448 0,500 0,792 0,890 |
В области разбавленных растворов (ниже 0,1 моль/л) коэффициенты активности зависят главным образом от концентрации и заряда ионов, присутствующих в растворе, и мало зависят от природы растворенных веществ. Эта закономерность известна в теории растворов под названием «правила ионной силы», учитывающее электростатическое взаимодействие ионов и диполей. Согласно этому правилу ионы одинакового заряда независимо от их природы в разбавленных растворах с одинаковой ионной силой имеют равные коэффициенты активности. Ионной силой раствора (I) называется полусумма произведений концентрации всех ионов, на квадрат их заряда (z):
 (11.8)
(11.8)
где i – порядковый номер иона.
3. Водородный показатель. Индикаторы. Вода служит не только наиболее распространенным растворителем для многих электролитов, но и сама является идеальным амфолитом. В соответствии с равновесием
H2O ⇄ H+ + OH–
в чистой воде присутствуют катионы водорода и гидроксид-анионы в строго эквивалентных количествах. Константа диссоциации воды
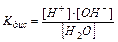 (11.9)
(11.9)
имеет значение в стандартных условиях – 1,8×10–16 т.е. вода диссоциирована в очень малой степени. Так как вода – очень слабый электролит, то концентрация недиссоциированных молекул может быть принята равной общему количеству молей в 1 л воды, т.е. [Н2O] = 1000/18 = 55,56 моль/л. Тогда Kдис×[H2O] = [H+]×[OH–] или [H+]×[OH–] = 1,8×10–16× 55,56 = 10–14 (константа воды).
Величина [H+]×[OH–] = 10–14 называется ионным произведением воды –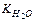 , Kw. Так как в воде концентрации гидратированных ионов равны, то [H+] = [OH–] =
, Kw. Так как в воде концентрации гидратированных ионов равны, то [H+] = [OH–] = 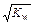 = 10–7 моль/л.
= 10–7 моль/л.
Константа равновесия Kw увеличивается с ростом температуры. При добавлении кислоты концентрация ионов водорода увеличивается и соответственно уменьшается концентрация гидроксид-ионов, поскольку при данной температуре ионное произведение воды – величина постоянная. При добавлении щелочи наблюдается обратная картина. Таким образом, концентрация ионов водорода в растворе может служить мерой кислотности или щелочности среды. В кислых растворах [H+] > 10–7, в щелочных [H+] < 10–7. Вводится значение десятичного логарифма концентрации водородных ионов с обратным знаком, которое называют водородным показателем pH:
pH = –lg[H+] (11.10)
Тогда для нейтральной среды pH = 7, для кислых растворов рН < 7, а для щелочных рН > 7. Аналогичным образом реакция среды может быть охарактеризована так называемым гидроксильным показателем:
pOH = –lg[OH–](11.11)
Для воды рН = рОН = 7, а изменение рОН в кислых и щелочных растворах противоположно изменению рН. Прологарифмировав ионное произведение воды, получим
lg[H+] + lg[OH–] = –14 (11.12)
Взяв логарифмы со знаком «–» , получим соотношение рН+рОН=14, Водородный показатель удобно представить в виде шкалы (рис. 11.2).
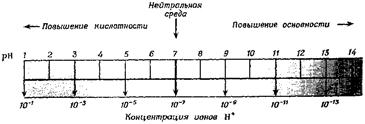
Рис. 11.2. Шкала pH.
Для определения рН используют так называемые кислотно-основные индикаторы – вещества, меняющие свой цвет в зависимости от относительной концентрации ионов H+ и ОН–. Индикаторы представляют собой слабые органические кислоты или основания. Одним из наиболее известных индикаторов является лакмус, окрашивающийся при избытке Н+ (т.е. в кислой среде) в красный цвет, при избытке ОН– (т.е. в щелочной среде) – в синий и имеющий в нейтральной среде фиолетовую окраску за счет равновесия: HInd ⇄ H+ + Ind–.
§ 4. Гидролиз. Равновесие с участием малорастворимых веществ. Наряду с электролитической диссоциацией и гидратацией (напомним, что это процесс взаимодействия ионов с молекулами воды за счет донорно-акцепторных связей, как правило, с образованием гидратированных ионов) имеется еще реакция гидролиза («разложения» водой). Гидролизом называется обменная реакция разложения соли водой, в результате которой получаются слабые кислоты или основания. Реакция гидролиза обратна реакции нейтрализации
Рассмотрим важнейшие случаи гидролиза солей.
1) Соль слабой кислоты и сильного основания. Сюда относятся такие соли как Na2CO3, K2CO3, CH3COONa, KCN и т.д.
Na2CO3 + HOH ⇄ NaHCO3 + NaOH
CO32– + HOH ⇄ HCO3–+ OH–
KOH сильное основание, хорошо диссоциируется в водном растворе, а CH3COOH – слабая кислота, распадающаяся на ионы лишь в очень малой степени. Раствор приобретает щелочную реакцию, вследствие наличия в нем свободных гидроксильных ионов в концентрации, более высокой, чем H+ ионов
[OH–]>[H+], pH>7.
2) Соль слабого основания и сильной кислоты. Сюда относятся NH4Cl, NH4NO3, AlCl3, CuSO4 и т.д.
NH4NO3 + HOH ⇄NH4OH + HNO3
NH4+ + HOH ⇄ NH4OH + H+
NH4OH – слабое основание, малодиссоциирующее, HNO3 – сильная кислота, сильнодиссоциирующая. [H+]>[OH–], pH < 7.
3) Соль образованная слабым основанием и слабой кислотой. Сюда относятся NH4CN, Al2S3,(CH3COO)3Fe
NH4CN + HOH ⇄ NH4OH+HCN
Раствор приобретает слабокислую, если кислота сильнее основания или слабощелочную, если основание сильнее кислоты, реакцию.
4) Соль, образованная сильным основанием и сильной кислотой. Она не гидролизуется, т.к. обратная гидролизу реакция нейтрализации практически необратима, т.е. протекает да конца.
Показателем глубины протекания процесса гидролиза служит степень гидролиза
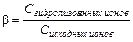 (11.13)
(11.13)
Гидролиз соли, образованной слабой кислотой HA и сильным основанием, характеризуется константой гидролиза KГ:
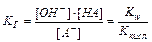 (11.14)
(11.14)
Kw – ионное произведение воды.
Аналогично для соли слабого основания MOH и сильной кислоты:
 (11.15)
(11.15)
Произведение растворимости. В насыщенном растворе малорастворимого сильного электролита устанавливается равновесие между осадком (тв. фазой) электролита и ионами электролита в растворе, например:
BaSO4 (тв.) ⇄Ba2+ (р-р) + SO42– (р-р)
Т.к. в растворах электролитов состояние ионов определяется их активностями, то константа равновесия процесса выразится уравнением
 (11.16)
(11.16)
Активность сульфата бария является при данной температуре константой. Следовательно, произведение активностей ионов Ba2+ и SO42– представляют собой постоянную величину, называемую произведением растворимости и обозначаемую ПР:
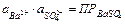 (11.17)
(11.17)
Произведение активностей ионов малорастворимого электролита, содержащихся в его насыщенном растворе (произведение растворимости), есть величина постоянная при данной температуре.
Если электролит очень мало растворим, то ионная сила его насыщенного раствора близка к нулю, а коэффициент активности ионов мало отличается от единицы. В подобных случаях произведение активностей ионов в выражении можно заменить произведением их концентраций
 ,
, 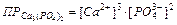
При увеличении концентрации одного из ионов электролита в его насыщенном растворе произведение концентраций ионов электролита становится больше ПР. При этом равновесие между твердой фазой и раствором смещается в сторону образования осадка. Таким образом, условием образования осадка является превышение произведения концентраций ионов малорастворимого электролита над его произведением растворимости.
Если в насыщенном растворе электролита уменьшится концентрация одного из ионов, произведение концентраций ионов будет меньше значения ПР, раствор станет насыщенном, а равновесие между жидкой фазой и осадком смещается в сторону растворения осадка. Следовательно, растворение осадка малорастворимого электролита происходит при условии, что произведения концентраций его ионов меньше значения ПР.
§ 5. Структура воды. Водородная связь.Промежуточный характер между валентным и межмолекулярным взаимодействием имеет так называемая водородная связь. Она осуществляется между положительно заряженным протоном (поляризованным атомом водорода), химически связанным в одной молекуле, и отрицательно заряженным ионом гетероатома (поляризованным атомом фтора, кислорода, азота или хлора), принадлежащим другой молекуле. То, что подобное взаимодействие не обнаруживается у других атомов, обусловлено уникальными свойствами поляризованного атома водорода, протона – его малым размером и отсутствием электронов. Водородная связь проявляется тем сильнее, чем больше относительная электроотрицательность и меньше размер атома-партнера.
Благодаря наличию водородной связи молекулы объединяются в димеры и более сложные ассоциаты, устойчивые в растворах. Ассоциаты (или кластеры) – многомолекулярные образования могут представлять собой одномерные образования, двумерные плоские сетки и трехмерные пространственные структуры.
В кристалле льда молекулы воды расположены тетраэдрически. Каждый атом кислорода тетраэдрически связан с четырьмя атомами водорода. Создается ажурная структура, далекая от плотной упаковки. При плавлении льда водородные связи частично разрушаются. Это сближает молекулы, поэтому вода плотнее льда.

| Рис. 11.3. Структура льда с водородными связями. |
Силы, обеспечивающие возможность существования жидкой фазы, носят различный характер. Чаще всего это межмолекулярные ван-дер-ваальсовые силы взаимодействия типа диполь-диполь или диполь-индуцированный диполь, либо дисперсионные, описанные Лондоном и называемые силами взаимодействия Ван-дер-Ваальса - Лондона. Энергия последних сильно зависит от расстояния между центрами взаимодействующих частиц (она пропорциональна 1/r6), поэтому такие взаимодействия могут возникать только на очень коротких расстояниях. Энергия межмолекулярных взаимодействий, как правило, на 2-3 порядка меньше энергии обычных химических связей (2-20 кДж/моль).
Одним из интересных типов межмолекулярного взаимодействия является водородная связь, наиболее ярко и своеобразно проявляющаяся в жидкой воде, придающая особый облик ее структуре и химическим свойствам. Именно наличием водородной связи обусловлены свойства воды как универсального растворителя.
В системах с наиболее прочными водородными связями межъядерные расстояния O-Н и Н-O во фрагменте O-Н...О выравниваются. Самая короткая водородная связь обнаружена в анионе НF2–, здесь она симметрична и имеет энергию связи порядка 155-242 кДж.
Еще одним примером проявления сильной водородной связи является катион Н5О2+(напомним существование Н3О+), реализующиеся в воде в форме димера [Н2О-Н-ОН2]+, существующий в воде при определенных значениях рН. В линейном фрагменте О-Н-О реализуется водородная связь, расстояние О-О равно 0,25 нм, расстояние O-Н – 0,125 нм, энергия связи примерно 150 кДж. На этом примере можно видеть, как в результате образования Н-связей резко сокращается расстояние между взаимодействующими частицами по сравнению с обычным межмолекулярным взаимодействием. Для ассоциатов, кластеров с большим числом молекул воды водородная связь приобретает ассиметричный характер: фрагмент О-Н-О длиной 0,36 нм имеет два неравных плеча О-Н – 0,1 и 0,26 нм.
Например, в структуре льда между атомами кислорода во фрагменте O-Н...О расстояние О...О составляет 0,276 нм, что заметно отличается от 0,36 нм, однако больше, чем 0,25 нм, характерного для очень сильной водородной связи.
Какова же природа водородной связи? Долгое время ее описывали как не совсем обычное электростатическое взаимодействие.
Более глубокое проникновение в природу взаимодействий показало несостоятельность электростатического подхода. Основой электростатического взаимодействия прежде всего является ее ненаправленный характер. Поэтому появление водородной связи следовало ожидать в любом пространственном расположении двух взаимодействующих молекул. Однако опыт показывает, что водородная связь имеет строго направленный характер. И, наконец, при электростатическом подходе невозможно объяснить перераспределение валентной электронной плотности при взаимодействии молекул, которое играет существенную роль при образовании водородной связи.
Полные квантовомеханические расчеты показывают, что в линейном фрагменте О-Н···О происходит общий сдвиг валентной электронной плотности от одного кислорода к другому.
Таким образом, по своей природе водородная связь является разновидностью донорно-акцепторной связи. Особенность ее заключается в том, что электронная пара атома О молекулы Н2О непосредственно взаимодействует с атомом Н группы О-Н, через которую и осуществляется перенос заряда на другую молекулу воды. В результате такого взаимодействия происходит увеличение дипольного момента комплекса с водородной связью по сравнению с геометрической суммой дипольных моментов изолированных молекул воды. Так, эффективный дипольный момент молекул воды, связанных водородными связями, равен 2,60 Д, а свободных молекул воды – 1,85 Д.
Чем больше молекул вовлекается в образование комплекса с водородными связями, тем в большей степени происходит перераспределение электронного заряда, тем сильнее водородные связи и выше их энергия. Это означает, что в жидких системах водородные связи носят кооперативный характер, они пронизывают всю жидкую систему, вовлекая в такое взаимодействие огромное число молекул.
Итак, водородная связь есть особый вид межмолекулярного донорно-акцепторного взаимодействия, в котором принимают участие все атомы, входящие в состав молекул. Это взаимодействие носит характер направленного дрейфа электронной плотности от молекулы-донора к молекуле-акцептору. Специфика водородной связи связана с уникальной электронной структурой атома водорода, поскольку в этом случае атом молекулы-донора со своей неподеленной электронной парой может приблизиться на очень короткое расстояние к атому Н, входящему в состав молекулы акцептора. Уникальность электронной структуры атома водорода заключается в отсутствии у него заполненных электронных оболочек. Специфика водородной связи в воде и водных растворах можно охарактеризовать двумя терминами: эстафетность и динамичность, т.е. протоны обеспечивают структурную организацию кластеров за счет большой скорости «эстафетного» переноса электронной плотности. Уместно сравнить явление с «бегущей волной» зрителей на стадионе, тем самым, подчеркивая волновой характер делокализованной химической связи для большого ансамбля числа молекул.
Более детально свойства водных растворов электролитов рассмотрены в дополнении к лекции 12.
Лекция 13
Окислительно-восстановительные реакции
Другим важным типом химических реакций, протекающих, как правило, в водных растворах, являются окислительно-восстановительные реакции или реакции с переносом электрона, в которых одни реагенты теряют, а другие приобретают электроны, меняют степень окисления.
§ 1. Степень окисления. Одним из основных понятий в химии, широко использующимся при составлении уравнений ОВР, является степень окисления (с.о.) атомов.
С.о. атома (элемента) в соединении – это условный заряд, вычисленный в предположении, что соединение состоит только из ионов. При определении с.о. условно предполагают, что валентные электроны в соединении переходят к более электроотрицательным атомам, а потому соединения состоят из положительно и отрицательно заряженных ионов. В действительности же в большинстве случаев происходит не полная отдача электронов, а только смещение электронной пары от одного атома к другому. Тогда можно дать другое определение: Степень окисления – это тот электрический заряд, который возник бы на атоме, если бы электронные пары, которыми он связан с другими атомами в соединении, перешли к более электроотрицательным атомам, а электронные пары, связывающие одинаковые атомы, были бы между ними поделены.
При вычислении степеней окисления используется ряд простых правил. С.о. простых веществ, как одноатомных, так и молекулярных, равна 0 (Fe0, O ). С.о. любого простого одноатомного иона равна заряду этого иона (Na+1, Ca+1, S–2). С.о. водорода в соединениях равна +1 (
). С.о. любого простого одноатомного иона равна заряду этого иона (Na+1, Ca+1, S–2). С.о. водорода в соединениях равна +1 ( ,
, ), за исключением гидридов металлов, где она равна -1 (
), за исключением гидридов металлов, где она равна -1 ( ,
, ). С.о. кислорода в соединениях равна -2 (
). С.о. кислорода в соединениях равна -2 (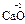 ,
,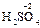 ); за исключением пероксидов, где она формально равна -1 (
); за исключением пероксидов, где она формально равна -1 (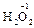 ), и фторида кислорода, где она равна +2 (
), и фторида кислорода, где она равна +2 (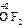 ). С.о. фтора в соединениях всегда равна -1, с.о. других галогенов (Cl, Br, I) равна -1, за исключением соединений с более электроотрицательными элементами, в которых она принимает положительные значения от +1 до +7 (
). С.о. фтора в соединениях всегда равна -1, с.о. других галогенов (Cl, Br, I) равна -1, за исключением соединений с более электроотрицательными элементами, в которых она принимает положительные значения от +1 до +7 (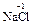 ,
, 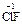 ,
,  ).
).
В ковалентных соединениях неметаллов более электроотрицательному элементу приписывается отрицательная с.о., равная заряду его наиболее распространенного аниона. Например, в CCl4 с.о. хлора -1, а углерода +4; в SF6 с.о. F
-1, а серы +6; но в CS2 с.о. серы -2, тогда с.о. углерода +4.
Алгебраическая сумма с.о. в нейтральной молекуле равна нулю, в комплексном ионе – заряду иона. Например, в NH4Cl сумма с.о. всех атомов водорода равна 4×(+1), а с.о. хлора -1, следовательно, с.о. азота должна быть -3. В сульфат-ионе SO42– сумма с.о. четырех атомов кислорода равна -8, поэтому сера должна иметь с.о. +6, чтобы полный заряд иона был равен -2. В химических реакциях должно выполняться правило сохранения алгебраической суммы с.о. всех атомов. В полном уравнении химической реакции окислительные и восстановительные процессы должны точно компенсировать друг друга.
Хотя с.о., как отмечалось выше, довольно формальное понятие, оно применяется в химии для следующих целей: во-первых, для составления уравнений ОВР, во-вторых, для предсказания окислительно-восстановительных свойств элементов в соединении.
Таблица 13.1
Степени окисления различных элементов
| Элемент | Значения степени окисления и примеры соединений |
| F | –1 (HF, KF) |
| O | –2 (H2O, CaO, CO2); –1 (H2O2); +2 (OF2) |
| N | –3 (NH3); –2(N2H4); –1 (NH2OH); +1 (N2O); +2 (NO); +3 (N2O3, HNO2); +4 (NO2); +5 (N2O5, HNO3) |
| Cl | –1 (HCl, NaCl); +1 (NaClO); +3 (NaClO2); +5 (NaClO3); +7 (Cl2O7, NaClO4) |
| Br | –1 (KBr); +1 (BrF); +3 (BrF3); +5 (KBrO3) |
| I | –1 (HI); +1 (ICl); +3 (ICl3); +5 (I2O5); +7 (IO3F, K5IO6) |
| C | –4 (CH4); +2 (CO); +4 (CO2, CCl4) |
| Si | –4 (Ca2Si); +2 (SiO); +4 (SiO2, H2SiO3, SiF4) |
| H | –1 (LiH); +1 (H2O, HCl) |
| S | –2 (H2S, FeS); +2 (Na2S2O3); +3 (Na2S2O4); +4 (SO2, Na2SO3, SF4); +6 (SO3, H2SO4, SF6) |
| Se, Te | –2 (H2Se, H2Te); +2 (SeCl2, TeCl2); +4 (SeO2, TeO2); +6 (H2SeO4, H2TeO4) |
| P | –3 (PH3); +1 (H3PO2); +3 (H3PO3); +5 (P2O5, H3PO4) |
| As, Sb | –3 (GaAs, Zn3Sb2); +3 (AsCl3, Sb2O3); +5 (H3AsO4, SbCl5) |
| Li, Na, K | +1 (NaCl) |
| Be, Mg, Ca | +2 (MgO, CaCO3) |
| Al | +3 (Al2O3, AlCl3) |
| Cr | +2 (CrCl2); +3 (Cr2O3, Cr2(SO4)3); +4 (CrO2); +6 (K2CrO4, K2Cr2O7) |
| Mn | +2 (MnSO4); +3 (Mn2(SO4)3); +4 (MnO2); +6 (K2MnO4); +7 (KMnO4) |
| Fe | +2 (FeO, FeSO4); +3 (Fe2O3, FeCl3); +4 (Na2FeO3) |
| Cu | +1 (Cu2O); +2 (CuO, CuSO4, Cu2(OH)2CO3) |
| Ag | +1 (AgNO3) |
| Au | +1 (AuCl); +3 (AuCl3, KAuCl4) |
| Zn | +2 (ZnO, ZnSO4) |
| Hg | +1 (Hg2Cl2); +2 (HgO, HgCl2) |
| Sn | +2 (SnO); +4 (SnO2, SnCl4) |
| Pb | +2 (PbO, PbSO4); +4 (PbO2) |
Для многих элементов характерно несколько значений с.о. (табл. 13.1), и, вычислив его с.о., можно предвидеть окислительно-восстановительные свойства: элемент в наибольшей отрицательной степени окисления может только отдавать электроны (окисляться) и быть восстановителем, в наибольшей положительной с.о. – только принимать электроны (восстанавливаться) и быть окислителем, в промежуточных степенях окисления – и окисляться, и восстанавливаться. Кроме того, и другие свойства соединений элемента в одной и той же с.о. оказываются похожими. Теперь рассмотрим, как степени окисления используются при составлении ОВР.
§ 2. Составление уравнений окислительно-восстановительных реакций (ОВР).Исходя из основных определений реакции с переносом электрона для составления материального баланса ОВР применяют так называемый метод полуреакций: разделение стадий окисления и восстановления на отдельные. При этом следует четко принимать, что эти две стадии одновременны и суть одного процесса («две стороны одной медали»).
В каждой из них учитывается материальный и зарядный балансы, затем они стехиометрически складываются с учетом равенства числа электронов «отдающихся» и «принимающихся» в полуреакциях. Проиллюстрируем это на следующем примере (в «Практикуме» этот материал разобран более детально).
Рассмотрим реакцию K2Cr2O7 с KI. По методу полуреакций поступают следующим образом.
Исключаем из уравнения реакции все ионы-наблюдатели, т.е. ионы, реально не принимающие участия в реакции (в нашем случае K+ и SO42–):
Cr2O72– + I– + H+ ® Cr3+ + I2 + H2O.
Составляем уравнения полуреакций, одна из которых включает Cr, а другая I:
Cr2O72– ® 2Cr3+
2I– ® I2
Сбалансируем число атомов в уравнении каждой полуреакции, добавив к ним H+ и H2O, если реакция протекает в кислой среде, либо H2O и OH–, если реакция протекает в щелочной среде:
Cr2O72– + 14H+ ® 2Cr3+ + 7H2O (атомы Cr, O и H сбалансированы),
2I– ® I2.
Сбалансируем заряды, добавляя необходимое число электронов:
Cr2O72– + 14H+ + 6 ® 2Cr3+ + 7H2O,
® 2Cr3+ + 7H2O,
2I– ® I2 + 2.
Обратите внимание, что входящее в сбалансированные уравнения полуреакций число электронов точно соответствует изменению с.о. (два атома Cr принимают 6 электронов, два атома I отдают 2 электрона).
Умножаем уравнения полуреакций на такие коэффициенты, чтобы число электронов, отданных восстановителем, было равно числу электронов, принятых окислителем (в нашем случае второе уравнение надо умножить на три):
Cr2O72– + 14H+ + 6® 2Cr3+ + 7H2O,
6I–® 3I2 + 6.
Складываем уравнения двух полуреакций и исключаем одинаковые частицы, содержащиеся в правой и левой части. Убеждаемся, что уравнение соответствует законам сохранения массы и заряда:
Cr2O72– + 6I– + 14H+ = 2Cr3+ + 3I2 + 7H2O.
Добавляем в уравнение ионы-наблюдатели:
K2Cr2O7 + 6KI + 7H2SO4 = 4K2SO4 + Cr2(SO4)3 + 3I2 + 7H2O.
Таким образом, кратко последовательность действий можно передать так: полуреакции–полная реакция–ионы–наблюдатели. Метод полуреакций наиболее удобен для составления уравнений реакций в водных растворах, особенно когда неизвестны все продукты реакции, однако для составления уравнений реакций с участием твердых веществ он неудобен, т.к. в этом случае часто приходится формально вводить в полуреакции не существующие на самом деле частицы, например, ионы O2–. При составлении уравнений электролиза также используется метод полуреакций, т.к. в этом случае полуреакции действительно происходят порознь, на разных электродах.
Видно, что ОВР в растворах – сложные; включают в себя реакции кислотно-основного равновесия (нейтрализация, гидролиз).
Среди ОВР выделяются реакции диспропорционирования, отличающиеся тем, что один и тот же элемент в одной и той же степени окисления является и окислителем, и восстановителем (поэтому в старых учебниках они назывались также реакциями самоокисления-самовосстановления):
2Pb2+ ⇄ Pb4++Pb0 2Cu+ ⇄ Cu2++Cu0.
Рассмотрим реакцию взаимодействия магния с разбавленной азотной кислотой, в результате которой получаются нитраты магния и аммония:
Mg + HNO3 ® Mg(NO3)2 + NH4NO3
Удалим из уравнения реакции ионы-наблюдатели
Mg + NO3– ® Mg2+ + NH4+.
Запишем уравнения полуреакций:
Mg ® Mg2+,
NO3– ® NH4+.
Сбалансируем атомы (обратите внимание, что среди продуктов второй
полуреакции добавляются три молекулы воды):
Mg ® Mg2+,
NO3– + 10H+ ® NH4+ + 3H2O.
Сбалансируем заряды:
Mg = Mg2+ + 2,
NO3– + 10H+ + 8= NH4+ + 3H2O.
Сложим полуреакции:
4Mg + NO3– + 10H+ = 4Mg2+ + NH4+ + 3H2O.
Добавим ионы-наблюдатели:
4Mg + 10HNO3 = 4Mg(NO3)2 + NH4NO3 + 3H2O.
Для многих ОВР состав продуктов зависит от условий протекания реакции. Так, при взаимодействии азотной кислоты с металлами в зависимости от природы металла и концентрации HNO3 последняя может восстанавливаться до аммиака (нитрата аммония), молекулярного азота, оксидов азота (II) или (IV), а на практике всегда получается смесь продуктов восстановления:
Ag + HNO3(конц.) = AgNO3 + NO2 + H2O
3Ag + 4HNO3(разб.) = 3AgNO3 + NO + 2H2O
Одним из наиболее широко используемых в лабораторной практике окислителей является перманганат калия KMnO4 (список распространенных окислителей и восстановителей приведен в табл. 13.2). В зависимости от условий проведения реакция восстановления протекает по-разному:
MnO4– +  = MnO42– (в щелочной среде),
= MnO42– (в щелочной среде),
MnO4– + 2H2O + 3= MnO2¯ + 4OH– (в нейтральной среде),
MnO4– + 8H+ + 5= Mn2+ + 4H2O (в кислой среде).
Таблица 13.2
Распространенные окислители и восстановители
| Окислители | Восстановители |
| Свободные галогены Кислород O2, озон O3, пероксид водорода H2O2 Азотная кислота HNO3, оксид азота (IV) NO2 Конц. серная кислота H2SO4 Селеновая кислота H2SeO4 Перманганат калия KMnO4, манганат калия K2MnO4, Оксид марганца (IV) MnO2 Бихромат калия K2Cr2O7, хромат калия K2CrO4 Оксид меди (II) CuO, оксид серебра (I) Ag2O Оксид свинца (IV) PbO2 Ионы благородных металлов (Ag+, Au3+) Хлорид железа (III) FeCl3 Гипохлориты, хлораты и перхлораты Царская водка (смесь HNO3+3HCl) Анод при электролизе | Свободные металлы Водород и гидриды металлов Уголь, оксид углерода (II) CO Сероводород H2S, оксид серы (IV) SO2 Сернистая кислота H2SO3 и ее соли Иодоводородная кислота HI и ее соли, бромоводородная кислота HBr, соляная кислота HCl Хлорид олова (II) SnCl2, сульфат железа FeSO4, сульфат марганца MnSO4 Азотистая кислота HNO2, аммиак NH3, гидразин N2H4, оксид азота (II) NO Фосфористая кислота H3PO3 Альдегиды, спирты, муравьиная и щавелевая к-ты Глюкоза Катод при электролизе |
§ 3. Окислительно-восстановительные эквиваленты.При рассмотрении взаимодействия кислот и оснований удобно использовать понятия кислотно-основного эквивалента и эквивалентной массы кислоты (основания). Эквивалентная масса кислоты равна молярной массе, деленной на основность кислоты. Для одноосновных кислот эквивалентная масса совпадает с молярной массой, например MЭ(HCl) = M(HCl) = 36,5 г/моль; для двухосновных кислот эквивалентная масса равна половине молярной массы (MЭ(H2SO4) = M(H2SO4)/2 =
(98 г/моль)/2 = 49 г/моль) и т.д. Таким образом, эквивалентная масса кислоты – это количество кислоты, которое способно отщепить 1 моль протонов. Аналогично, эквивалентная масса основания – это количество основания, которое способно нейтрализовать 1 моль протонов (или отщепить 1 моль гидроксид-ионов), например, MЭ(NaOH) = M(NaOH) = 40 г/моль; MЭ(Ca(OH)2) = M(Ca(OH)2)/2 =
(74 г/моль)/2 = 37 г/моль.
Теперь аналогичным образом введем определение окислительно-восстановительного эквивалента.
Эквивалентом окислителя (восстановителя) называется такое количество вещества, которое в ходе ОВР принимает (отдает) 1 моль электронов.
Эквивалентную массу окислителя (восстановителя) легко найти, разделив молярную массу на число принимаемых (отдаваемых) в реакции электронов. Например сернистая кислота, отдавая два электрона, окисляется в серную, тогда эквивалентна масса равна
MЭ(H2SO3) = M(H2SO3)/2 = (82 г/моль)/2 = 41 г/моль.
Обратите внимание, что для многих веществ эквивалентная масса – величина переменная, зависящая от того, что какой реакцией она определяется. Так, перманганат калия в различных условиях (см. выше) может принимать один, три или пять электронов, и эквивалентная масса для этих случаев будет составлять соответственно M(KMnO4), M(KMnO4)/3, M(KMnO4)/5 (158 г/моль, 52,7 г/моль, 31,6 г/моль). Когда на склянке с 0,02 М раствором KMnO4 пишут 0,1 н. р-р KMnO4, то при этои подразумевают, что все реакции с его участием будут происходить в кислой среде, MnO4–восстановится до Mn2+. Если применять этот раствор в других условиях, то использование его нормальности в расчетах приведет к ошибке.
Раствор KMnO4 точно известной концентрации используют для определения концентрации восстановителей. Этот метод анализа называется перманганатометрией.
Ещё одна важная для химического анализа реакция – взаимодействие иода с тиосульфатом натрия, происходящее согласно уравнению:
I2 + 2Na2S2O3 = 2NaI + Na2S4O6.
Эта реакция может использоваться для определения содержания иода, но чаще иодометрия используется для определения концентрации окислителей.
§ 4. Потенциал восстановления (окисления). Разность потенциалов.Учитывая, что окислительно-восстановительные реакции протекают с переносом электрона – заряда, то каждая стадия (полуреакция) обладает электростатическим потенциалом (Е, j), выражаемых в вольтах (В). В природе потенциалы окисления, восстановления меняются практически от –3 до +3 В. Чтобы не путаться в знаках потенциала, принято писать их в форме потенциалов восстановления, т.е. +. Все они табулированы (см. «Практикум») в такой форме. Для ОВР соответственно вводится разность потенциалов (DЕ, Dj) – ЭДС, В. Разность потенциалов ОВ системы, с точки зрения термодинамики, связана с DG соотношением: DG = –Amax = nFDE, где Amax – работа, совершаемая системой, n – число электронов, F – число Фарадея 96484,56 Кл/моль. Таким образом, разность потенциалов (DE) является термодинамической характеристикой ОВР, и следуя выше приведенному уравнению реакция будет протекать самопроизвольно (слева направо), когда DE > 0 – положительна. Таким образом, разность потенциалов характеризует направленность ОВР. В «Практикуме» приведены примеры и лабораторные работы, иллюстрирующие это положение. На практике способность ОВ систем за счет разности потенциалов производить полезную работу реализуется в гальванических элементах (Вольта, Якоби-Даниэля), батарейках и др.
Количественно равновесный (реальный) потенциал связан с природой и концентрацией ОВ пары по формуле Нернста:
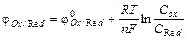 , (13.1)
, (13.1)
где jOx/Red – равновесный потенциал; – стандартный ОВ потенциал (он табулирован и определяется только природой веществ); R – универсальная газовая постоянная; Т – температура, К; n – число электронов; COx/Red – концентрации окислителя (восстановителя), моль/л.
– стандартный ОВ потенциал (он табулирован и определяется только природой веществ); R – универсальная газовая постоянная; Т – температура, К; n – число электронов; COx/Red – концентрации окислителя (восстановителя), моль/л.
При подстановке в формулу Нернста значений R, Т = 298 К для стандартных условий и коэффициента перевода натурального логарифма в десятичный (2,303) и она будет:
 . (13.2)
. (13.2)
ОВР протекают как в свободном объеме, так и на электродах, что мы рассмотрим в следующих лекциях.
§ 5. Диаграмма электрохимической устойчивости воды. Химическая стойкость металлов в водных растворах. Металл, растворяющийся в воде, растворе кислоты или основания, является восстановителем (Red), он окисляется. Это растворение может быть целенаправленным (химическое фрезерование) и нежелательным (коррозия). Необходимым условием протекания окислительно-восстановительного (Ox-Red) процесса является наличие окислителя (Ox). С учетом протекания ОВР в воде, то необходимо рассмотреть роль воды в них. Напомним, что в воде имеются Н+, OH–и растворенный О2.
В кислотной среде (0£рН<7) в качестве окислителя могут выступать ионы водорода Н+ или «сложный» окислитель Н++О2; в нейтральной (рН=7) и в щелочной среде (7<рН£14) окислителем являются молекулы H2O или смесь воды и кислорода (H2O+O2), а также OH–.
Металл, контактирующий одновременно с газом и раствором, содержащим ионы этого газа, называется газовым электродом (водородным, кислородным и др.). Потенциалы водородного и кислородного электродов не зависят от формы разряжающихся частиц (молекул воды или ионов, на которые она диссоциирует), но зависят от рН среды и парциального давления водорода (PH2) и кислорода (PO2):
2Н++2 =Н2 2H2O+2=2ОН–+Н2
=Н2 2H2O+2=2ОН–+Н2
4Н++О2+4=2H2O 2H2O+О2+4=4ОН–
Зависимость jН+/Н2и jО2/ОН–от рН при PH2= PO2= 1ат представлена уравнениями и графически – в виде диаграммы электрохимической устойчивости воды (рис. 13.1).
jН+/Н2=–0,059 рН, jО2/ОН– = 1,23–0,059 рН
По диаграмме (рис. 13.1) можно установить химическую стойкость металлов в растворах с различным значением рН. Отметим, что известный ряд напряжений jMen+/Me, B:
| Li+/Li | K+/K | Na/Na | Mg2+/Mg | Al3+/Al | Zn2+/Zn | Fe2+/Fe | ||||||
| –3,02 | –2,92 | –2,71 | –2,38 | –1,66 | –0,76 | –0,44 | ||||||
| Pb2+/Pb | 2H+/H2 | Cu2+/Cu | Ag+/Ag | Pt2+/Pt | Au+/Au | |||||||
| –0,13 | 0,00 | +0,34 | +0,79 | +1,20 | +1,70 | |||||||
изначально был известен как ряд способности замещения металлов друг другом в водных растворах («вытеснительный» ряд Бекетова). Когда же были установлены потенциалы Red/Ox пар металлов и катионов, он с точностью совпал с рядом Бекетова и получил количественную характеристику. С учетом электрохимической диаграммы воды он справедлив только при рН=0 (не учитывает влияние кислорода).
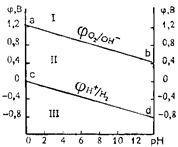 Рис. 13.1. Диаграмма электрохимической устойчивости воды.
Рис. 13.1. Диаграмма электрохимической устойчивости воды.
| Если стандартный потенциал металла (jМеn+/Me) положительнее jО2/ОН– (область 1, выше линии ab), то растворение металла с процессами восстановления по уравнениям невозможно. Все точки в области 1 соответствуют состоянию когда молекулы воды (или ионы гидроксила) могут выступать лишь как восстановители. Такое состояние имеет место на аноде при электролизе водных растворов или при взаимодействии воды или основания с сильным окислителем: |
2Cl2+2H2O=4HCl+O2, 2Cl2+4NaOH=4NaCl+2H2O+O2.
В этих реакциях молекулы воды и ионы гидроксила выступают как восстановители. Если jМеn+/Me положительнее jН+/Н2и отрицательнее jО2/ОН–(все точки между линиями ab и cd, область II), то растворение металла возможно, когда окислителем является (H++O2) или (H2O+O2), а не ионы водорода или молекулы воды. Все точки в области II соответствуют электрохимической устойчивости воды. В этой области в химических реакциях и при электролизе вода не может выступать ни окислителем, ни восстановителем.
Все точки ниже линии cd (область III) отвечают состоянию системы, когда молекулы H2O или Н+ в реакциях с металлами выступают как окислители. Этот случай условно называется «коррозией с водородной деполяризацией», которая происходит при взаимодействии H2O или иона Н+ с активными металлами. В области III при наличии в растворе газообразного кислорода в качестве окислителя в реакции с металлами могут также выступать сложные окислители (H2O + O2) или (H+ + O2). Этот случай условно называется «коррозией с кислородной деполяризацией». Таким образом, в области III могут выступать четыре окислителя, т.е. имеется возможность протекания четырех реакций восстановления.
В общем случае, при наличии в растворе нескольких видов ионов или недиссоциированных молекул электрохимически активных веществ последовательность протекания реакций восстановления определяется величиной их стандартного потенциала. В первую очередь, окислителем выступают те ионы, молекулы или их сочетания, которые характеризуются наиболее положительным потенциалом. Это, в частности, имеет место при растворении металлов в кислородсодержащих кислотах (HNO3, H2SO4 и др.), когда в качестве окислителя могут выступать анионы кислотного остатка.
Исходя из изложенного видно, что металлы находящиеся ниже линии водородного электрода встречаются, как правило, в соединениях в катионной форме; металлы находящиеся выше линии кислородного электрода встречаются в природе в форме самородков (золото).
Учитывая сказанное, легко объяснить, почему при взаимодействии металлов с азотной кислотой не выделятся газообразный водород. Примеры приведены в «Практикуме».
Лекция 14
Электрохимические процессы и системы
Процессы взаимного превращения химической и электрической форм энергии, протекающие на электродах (Ox/Red) реакции называются электрохимическими, их можно рассматривать как:
1. Самопроизвольные процессы, при которых химическая энергия превращается в электрическую. Сюда относятся химические источники тока (гальванические элементы), а также коррозионные процессы.
2. Электролиз – несамопроизвольный процесс, происходящий под действием внешнего источника постоянного электрического тока, при котором на электродах образуются новые вещества.
Особенности электрохимических процессов в том, что все они происходят на границе раздела двух фаз – твердая–жидкая – это электрод-электролит и являются Ox/Red реакциями. Ox/Red процессы определяются электродным потенциалом Ox/Red пары на электроде, напомним что на аноде идет процесс окисления, на катоде наоборот – восстановление, ЭДС процесса определяется как разность между 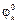 и
и 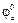 и его величина зависит от местоположения металлов в ряду электрохимической активности (напряжений).
и его величина зависит от местоположения металлов в ряду электрохимической активности (напряжений).
§ 1. Электродный потенциал металла. Двойной электрический слой. Электродом (первого рода) называют любой металл (или токопроводящий материал), погруженный в раствор электролита. Между ними существует подвижное равновесие, которое можно выразить уравнением Me ⇄ Men++n.
При погружении же металла в раствор начинается сложное взаимодействие металла с компонентами раствора электролита. Если энергия кристаллической решетки металла (Екрист) меньше энергии гидратации (Егидр) – Екрист < Егидр, то образующиеся ионы металла гидратируются и переходят в раствор, на поверхности металла устанавливается равновесие: Me+mH2O ⇄ Men+·mH2O+n. При этом скорости прямого и обратного переходов ионов будут равны. Процесс перехода атомов металла электрода в раствор связан с потерей электронов и образованием положительно заряженных ионов, т.е. происходит окисление (Me – n®Men+), обратный процесс – восстановление Men++n®Me0.
При равенстве скоростей прямого и обратного процессов uок = uвосст электрод находится в равновесном состоянии. Положение равновесия зависит как от энергии ионизации атома металла, так и от концентрации его ионов в растворе. Гидратированные ионы металла, находящиеся у его поверхности и оставшиеся на металле избыточные электроны образуют две противоположно заряженные плоскости, аналогичные обкладке плоского конденсатора – возникает двойной электрический слой (рис. 14.1). Между металлом и раствором возникает разность потенциалов, которая называется электродным потенциалом или потенциалом электрода.
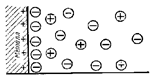 Рис. 14.1. Схема двойного электрического слоя на границе раздела металл/раствор.
Рис. 14.1. Схема двойного электрического слоя на границе раздела металл/раствор.
| 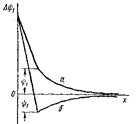
|
Часть двойного электрического слоя, которая образуется непосредственно прилегающими к электроду ионами, называется плотной частью двойного слоя (слой Гельмгольца). Толщина плотного слоя равна радиусу гидратированных ионов. В этой части имеет место линейное изменение потенциала между электродом и плоскостью, проходящей через центры прилегающих к нему ионов. Часть двойного электрического слоя, расположенная за плотной частью, называется диффузной (слой Гуи-Чапмена). Изменение потенциала в ней нелинейно. Полный скачок потенциала на границе электрода с электролитом слагается из суммы скачков потенциалов в плотной и диффузной частях двойного электрического слоя. В 1924 г. Штерн предложил адсорбционную теорию двойного электрического слоя. Он полагал, что определенная часть ионов удерживается вблизи поверхности раздела металл-электролит, образуя плотную гельмгольцевскую обкладку двойного слоя, с толщиной, отвечающей среднему радиусу ионов электролита, остальные ионы распределяются диффузно с постепенно убывающей плотностью заряда. Дальнейшее развитие теории строения двойного электрического слоя было дано в работах Фрумкина и его школы. По их мнению, в плотной части двойного слоя следует различать внутренний и внешний гельмгольцевские слои. Внутренний слой образован специфически адсорбированными ионами, частично или полностью дегидратированными и образующими диполи с металлом, во внешнем плотном слое находятся гидратированные ионы, притянутые к поверхности металла электростатическими слоями. Непосредственно за внешним гельмгольцевским следует диффузный слой.
Переход ионов металла с электрода в раствор приводит к равновесному состоянию системы. Потенциал, устанавливающийся в условиях равновесия ( ), называется равновесным электродным потенциалом и зависит от свойств металла, концентрации его ионов в растворе. Абсолютные значения электродных потенциалов определить экспериментально невозможно. Поэтому пользуются относительными значениями электродных потенциалов. Для этого находят разность потенциалов измеряемого электрода и электрода, потенциал которого условно принимают равным нулю.
), называется равновесным электродным потенциалом и зависит от свойств металла, концентрации его ионов в растворе. Абсолютные значения электродных потенциалов определить экспериментально невозможно. Поэтому пользуются относительными значениями электродных потенциалов. Для этого находят разность потенциалов измеряемого электрода и электрода, потенциал которого условно принимают равным нулю.
В настоящее время за нуль отчета принят потенциал стандартного водородного электрода. Он состоит из платинированной платины, контактирующей с
газообразным водородом и раствором HCl с СН+=1 моль/л, р = 1атм, T = 298 К (25°С), рН = 0. Возникает скачок потенциала за счет реакции 2Н++2⇄ Н2 между платиной и раствором.
Более подробно процессы, происходящие на платиновом электроде, можно представить схемой:
H2 (г.) ⇄ H2 (адс. Pt) ⇄ 2H (адс. Pt) 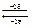 2H+ (адс. Pt) ⇄ 2H+ (р-р)
2H+ (адс. Pt) ⇄ 2H+ (р-р)
Обычно промежуточные стадии при кратком описании не приводят, но нужно помнить, что без развитой поверхности черненого платинового электрода равновесие между газовой фазой и раствором устанавливалось бы слишком долго.
Здесь уместно напомнить уравнение Нернста:
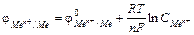 (14.1)
(14.1)
Стандартным электродным потенциалом металла 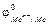 называют потенциал, который возникает на границе между металлом и раствором его соли с концентрацией ионов металла
называют потенциал, который возникает на границе между металлом и раствором его соли с концентрацией ионов металла 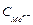 =1 моль/л при стандартных условиях, эта величина табулирована. Это количественная характеристика способности металла окисляться или восстанавливаться.
=1 моль/л при стандартных условиях, эта величина табулирована. Это количественная характеристика способности металла окисляться или восстанавливаться.
 измеряется относительно стандартного водородного электрода. Таким образом измерены стандартные потенциалы для всех металлов и они расположены в порядке возрастания величин этих потенциалов. Ряд стандартных электродных потенциалов называется рядом напряжений или рядом электрохимической активности металлов. Исходя из этого ряда можно сделать выводы: чем меньше , тем выше активность металла к окислению, а его ионам труднее восстанавливаться. Чем больше , тем металл труднее окисляется, а ионы легче восстанавливаются.
измеряется относительно стандартного водородного электрода. Таким образом измерены стандартные потенциалы для всех металлов и они расположены в порядке возрастания величин этих потенциалов. Ряд стандартных электродных потенциалов называется рядом напряжений или рядом электрохимической активности металлов. Исходя из этого ряда можно сделать выводы: чем меньше , тем выше активность металла к окислению, а его ионам труднее восстанавливаться. Чем больше , тем металл труднее окисляется, а ионы легче восстанавливаются.
В электрохимии используются металлические и газовые электроды.
Металлический электрод. При погружении металла в раствор собственных ионов устанавливается равновесие Me ⇄ Men++n. Для его измерения нужен гальванический элемент H2 Pt|H+||Men+|M. Токообразующей в этом элементе будет реакция Men++ H2 ⇄ Me+nH+. Скачок потенциала на границе Ме/электролит определяется по уравнению (14.1). Если электрод инертный (графитовый, угольный, Pt), то он является только передатчиком электронов, но в реакции сам не участвует.
H2 ⇄ Me+nH+. Скачок потенциала на границе Ме/электролит определяется по уравнению (14.1). Если электрод инертный (графитовый, угольный, Pt), то он является только передатчиком электронов, но в реакции сам не участвует.
Газовые электроды состоят из металлического проводника, контактирующего одновременно с газом и раствором, содержащим ионы этого газа. Металлический проводник служит для отвода и подвода . Наиболее часто используются водородный и кислородный газовые электроды. Скачок потенциала водородного электрода возникает на границе Н2/Н+ и потенциалоопределяющей является реакция 2H++2⇄ H2, выражающая равновесие на водородном электроде. 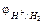 в нестандартных условиях можно рассчитать по уравнению (10.1), используя парциальное давление вместо концентрации водорода и при
в нестандартных условиях можно рассчитать по уравнению (10.1), используя парциальное давление вместо концентрации водорода и при  =1 моль/л, рН=0.
=1 моль/л, рН=0.  определяется по формуле =–0,059рН.
определяется по формуле =–0,059рН.
Аналогично водородному можно создать и кислородный электрод. Для этого необходимо Pt пластинку привести в контакт с O2 и раствором, содержащим ионы ОН–. Скачок потенциала происходит на границе О2/ОН– и протекают процессы 4Н+ + О2 + 4⇄ 2H2O, 2H2O + О2 + 4⇄ 4ОН–, потенциал определяет по уравнению; 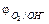 = 1,23 – 0,059 рН.
= 1,23 – 0,059 рН.
§ 2. Химические источники тока.Химические источники тока (ХИТ) представляют устройства, в которых химическая энергия Ox/Red реакции превращается в электрическую. ХИТы по принципу действия делятся на первичные – гальванические элементы, и вторичные – аккумуляторы.
Для получения электрического тока Ox/Red процессы должны быть пространственно разделены и система замкнута. Процесс окисления протекает на аноде (на электроде с более электроотрицательным потенциалом), восстановление – на катоде (на электроде с более электроположительным потенциалом). Название электродов определяются протекающими на них процессами.
ЭДС ХИТ равна разности потенциалов катода – (jk) и анода – (ja). При отсутствии тока в цепи: ЭДС = jk0 – ja0.
В процессе работы ХИТа его ЭДС может уменьшаться, что вызвано изменением потенциалов катода и анода при протекании электрического тока из-за поляризации электродов. Процесс уменьшения поляризации называется деполяризацией.
jk0, ja0 – потенциалы электродов без тока; 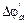 – смещение потенциала анода в положительную область;
– смещение потенциала анода в положительную область; 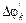 – смещение потенциала катода в отрицательную область под током. ЭДС0 = jk0 – ja0; ЭДС’=
– смещение потенциала катода в отрицательную область под током. ЭДС0 = jk0 – ja0; ЭДС’=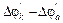 ; Епол=Dja+Djk. Напряжение гальванического элемента U<ЭДС из-за поляризации электродов и омических потерь. Напряжение ХИТ или напряжение на клеммах при электролизе представляет алгебраическую сумму:
; Епол=Dja+Djk. Напряжение гальванического элемента U<ЭДС из-за поляризации электродов и омических потерь. Напряжение ХИТ или напряжение на клеммах при электролизе представляет алгебраическую сумму:
U = ±[(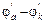 )+I(r1+r2)] (14.2)
)+I(r1+r2)] (14.2)
Знак «–» перед уравнением относится к ХИТ, а «+» к электролизу, r1 и r2 – сопротивление проводников 1-го и 2-го рода. При замыкании цепи I = 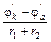 значительно меньше, чем I1 =
значительно меньше, чем I1 =  , r1+r2=const. Следовательно основная причина неравенства I<I1 вызвана изменением jk и ja при прохождении тока. Подробно гальванические элементы рассмотрены в «Практикуме». Здесь же мы проиллюстрируем как записывается на «электрохимическом языке» работа и устройство элемента Якоби–Даниэля.
, r1+r2=const. Следовательно основная причина неравенства I<I1 вызвана изменением jk и ja при прохождении тока. Подробно гальванические элементы рассмотрены в «Практикуме». Здесь же мы проиллюстрируем как записывается на «электрохимическом языке» работа и устройство элемента Якоби–Даниэля.
А Zn½ZnSO4½½CuSO4½Cu К
CuSO4 + Zn = ZnSO4 + Cu; Cu2++ SO42– + Zn0 = Zn2+ + SO42– + Cu0
окис: Zn – 2® Zn2+
восcт: Cu2+ + 2® Cu
Cu2+ + Zn0 = Zn2+ + Cu0
Отметим дополнительно, как устроен концентрационный гальванический элемент (у которого электроды одни и те же, но концентрация электролита в при электродном пространстве разная).
Ag (I)½AgNO3½½AgNO3½Ag (II) C1<<C2
А.: Ag – ® Ag+ (I)
К.: Ag+ + ® Ag0 (II)
Нетрудно убедиться, используя уравнение Нернста, что возникнет разность потенциалов.
Условия, необходимые для построения любого гальванического элемента: 1) наличие проводников 1-го рода (металлы) и 2-го рода (электролиты) (электронная и ионная проводимость); 2) возможность разделения системы электродов на две неравноценные части, т.е. разделены мембраной или электролитическим ключом.
Работа и устройства гальванических элементов, а также аккумуляторов и топливных элементов приведены в «Практикуме». Отметим лишь, что в аккумуляторах возможно протекание двух противоположно направленных процесса: выработка электрической энергии за счет окислительно-восстановительной реакции, протекающей на электродах, или разрядка и противоположный – зарядка, а по сути – электролиз. Должно быть понятным, что необходимым условием работы аккумулятора является обратимость процесса, чему удовлетворяют реакции диспропорционирования(например, 2PbSO4 + 2H2O ⇄ PbO2 + Pb + 2H2SO4) и неизменность концентрации ионов на электродах, последнее достигается когда ионы входят в состав труднорастворимых соединений.
Топливные элементы (электрохимические генераторы), в основе работы которых лежит реакция окисления топлива на электродах. Простейший и самый значимый топливный элемент основан на реакции окисления водорода кислородом: 2H2 + O2 = 2H2O. Его схему работы можно записать
H2, M½KOH½M, O2
M – проводник первого рода (токоотвод).
A.: 2ОН–+Н2 – 2= 2H2O
K.: 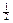 О2 + H2O + 2= 2ОН–
О2 + H2O + 2= 2ОН–
H2 + ½O2 = 2H2O
В результате протекания этой реакции в цепи генерируется постоянный ток и химическая энергия превращается в электрическую.
§ 3. Электролиз. ОВР, протекающие на электродах при прохождении через электролит электрического тока – электролиз. Для осуществления электролиза необходимы: 1) источник постоянного тока (стационарный электролиз);
2) электролит – водный раствор или расплав; 3) электроды.
Электрод, соединенный с отрицательным полюсом источника тока называется катод, на нем идет процесс восстановления (присоединения электронов), находящихся в растворе ионов металла или ионов водорода (как восстановление воды). Катод является лишь передатчиком электронов. Электрод, соединенный с положительным полюсом источника тока, называется анодом, на нем происходит процесс окисления (отдача электронов). Аноды бывают: растворимые и нерастворимые, инертные, они служат для отвода электронов. В качестве нерастворимых анодов используются графит, уголь, Pt. На аноде: выделяется анион электролита (2Cl–– 2® Cl2), окисление воды (2H2O – 4® О2 + 4Н+), или ионов гидроксила (4ОН– – 4® 2H2O + О2).
Количественные законы электролиза были установлены Фарадеем.
Первый закон. Масса веществ, выделившихся при электролизе на электродах, пропорциональна количеству прошедшего через раствор электричества и не зависит от его концентрации и температуры
m = k×q или m = k×I×t (14.3)
где m – масса металла, г; k – электрохимический эквивалент; I – сила тока, А;
t – время электролиза, с; q – количество электричества = I×t. Электрохимическим эквивалентом металла называется его масса, выделяющаяся на электроде при прохождении 1 кулон электричества, и характеризует природу металла.
Второй закон. Равные количества электричества выделяют при электролизе массы веществ, пропорциональные их химическим эквивалентам:
 , тогда объединенный закон:
, тогда объединенный закон: 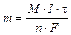 (14.4)
(14.4)
где М– молекулярная масса; n– валентность металла; хэ = М/n;
F – число Фарадея.
Из объединенного закона Фарадея следует, что nF– количество электричества, необходимое для выделения 1 моль атома металла или генерируемое гальваническим элементом при растворении такого же количества металла. Работа электрического тока (А), генерируемого в ХИТ при растворении 1 моль металла, равна А = nFE, где E = ЭДС ХИТ.
При расчетах электролиза вводится понятие плотность тока i – это отношение силы тока I к площади образца S
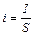 (14.5)
(14.5)
Наименьшее напряжение, при котором начинается процесс электролиза, называется напряжением разложения (U) данного электролита при данных условиях. Теоретически для электролиза достаточно было бы напряжения, равного ЭДС ХИТ., составленного из этих электродов, но имеющего противоположное направление, но на практике напряжение разложения больше, чем ЭДС ХИТ. Разность между напряжением, фактически необходимым для электролиза, и ЭДС ХИТ, называется перенапряжением (поляризацией) при электролизе (h)
h = U – ЭДС (14.6)
где U – напряжение разложения; ЭДС – ХИТ, отвечающего этой системе.
Выделительным потенциалом называется тот минимальный потенциал, при котором начинается фактический разряд ионов, который на величину перенапряжения превышает равновесный потенциал.
Любая электрохимическая реакция протекает минимум в 3 стадии: а) подвод реагентов к электроду; б) собственно электрохимическая реакция; в) отвод продуктов реакции от электродов.
Возникновение поляризации обусловлено замедленностью отдельных стадий процесса. В зависимости от характера замедленной стадии возникает концентрационная или электрохимическая поляризация. Концентрационная поляризация вызвана разностью концентраций ионов в объеме и приэлектродном слое (а, б – стадии). Изменение потенциала, обусловленное замедленностью электрохимической реакции (б), называется электрохимической поляризацией или перенапряжением – это превышение потенциала разряда ионов над нормальным потенциалом в равновесных условиях. Количественно связь между h и i выражается формулой Тафеля:
h = ±(а + bi) (14.7)
где h – перенапряжение; a, b – константы, зависят от природы материала, температуры.
Наиболее полно h изучено для ионов Н+ h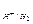 и отвечает процессу 2Н++2=Н2 на катоде; jк смещается в (–)-область под током. Перенапряжению кислорода h
и отвечает процессу 2Н++2=Н2 на катоде; jк смещается в (–)-область под током. Перенапряжению кислорода h отвечает процесс 4ОН––4®2H2O+О2 и jа' смещается в (+) область.
отвечает процесс 4ОН––4®2H2O+О2 и jа' смещается в (+) область.
§ 4. Электродные процессы при электролизе. Катодные процессы. Последовательность катодных процессов диктуется закономерностями Ox/Red реакций. На катоде первоначально разряжается тот ион, который характеризуется наиболее электроположительным потенциалом, т.е. наиболее сильные окислители. Все окислители условно можно разделить на 3 группы в соответствии с расположением металлов в ряду электрохимической активности j0 стандартных потенциалов. При электролизе водных растворов солей металлов, расположенных правее водорода (jМе>j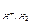 ) на катоде восстанавливаются ионы металла (Cu2++2®Cu), Вторая группа металлов расположена левее H2 до Mn, на катоде возможно одновременное восстановление и Н2 (или Н2О), и иона металла. Здесь необходимо учитывать hи зависимость jот рН. Третья группа металлов расположена левее Mn, при электролизе их можно получить только из расплавов.
) на катоде восстанавливаются ионы металла (Cu2++2®Cu), Вторая группа металлов расположена левее H2 до Mn, на катоде возможно одновременное восстановление и Н2 (или Н2О), и иона металла. Здесь необходимо учитывать hи зависимость jот рН. Третья группа металлов расположена левее Mn, при электролизе их можно получить только из расплавов.
Последовательность анодных процессов при электролизе также диктуется закономерностями ОВР. Возможны следующие реакции:
1) растворение металла Me – n® Men+;
2) образование оксидных покрытий Me2+ + 2ОН– ® MeО¯+ Н2О;
3) окисление анионов или Н2О.
На аноде первоначально протекает процесс, который характеризуется наименьшей величиной потенциала. При электролизе водных растворов с нерастворимым анодом необходимо руководствоваться рядом окисления анионов. При электролизе солей водных растворов бескислородных кислот разряжаются ионы I–, Br–, Cl–, а не молекулы Н2О. При электролизе солей, содержащих анионы NO3–, CO42–, PO43– на аноде окисляются молекулы Н2О или ОН–из-за высокого перенапряжения кислороды (h).
Рассмотрим электролиз водного раствора NaCl, рН=7. Анод нерастворимый.
К.: 2H2O + 2®H2+2OH–
А.: 2Cl–– 2®Cl2 (потенциал меньший), 2OH– – 2®H2O+O
При электролизе расплава NaCl (графитовые электроды) идет разложение NaCl = Na+ + Cl–, под током:
К.: Na– + ® Na0 – восстановление
А.: 2Cl– – 2® Cl2 – окисление.
Значение электролиза трудно переоценить, он имеет многочисленные и значительные прикладные аспекты (прикладная электрохимия): гальванотехника, гальваностегия, гальванопластика, электроэкстракция и др. Но это является предметом отдельного курса.
Лекция 15
КОРРОЗИЯ МЕТАЛЛОВ И ЗАЩИТА ОТ КОРРОЗИИ
Технически нецелесообразное разрушение металлов под воздействием внешней среды называется коррозией. Причиной коррозии металлов является их термодинамическая неустойчивость, т.е. возможность самопроизвольного перехода металла в более устойчивое окисленное состояние, в котором он встречается в природе. Химическая энергия реакции коррозионного разрушение металлов выделяется в виде теплоты и рассеивается в окружающем пространстве. Коррозионные процессы протекают необратимо в соответствии со вторым началом термодинамики и являются ОВ-процессами.
Убытки от коррозии делятся на: а) прямые, т.е. стоимость металла, подвергшегося коррозионному разрушению; б) косвенные, которые значительно могут превышать прямые потери.
К убыткам от коррозии можно отнести стоимость потерянного продукта, например, газа, нефти, масла через прокоррозировавшие трубы, экологический урон и т.д. Ежегодные потери от коррозии исчисляются многими миллиардами рублей.
По условиям протекания различают следующие виды коррозии:
1) газовая (при высоких температурах);
2) коррозия в неэлектролитах (органических средах, не проводящих электрический ток);
3) атмосферная коррозия в естественной атмосфере воздуха;
4) коррозия в электролитах (кислотная, щелочная, солевая, морская, в расплавах);
5) почвенная коррозия (ржавление подземных трубопроводов);
6) структурная коррозия из-за структурной неоднородности металла;
7) биокоррозия при участии продуктов, выделяемых микроорганизмами;
8) радиационная коррозия;
9) контактная коррозия, вызванная контактом металлов, имеющих разные потенциалы в данном электролите;
10) коррозионная кавитация – разрушение металла при одновременном коррозионном и ударном воздействии внешней среды (разрушение лопаток гребных винтов морских судов).
По характеру коррозионного разрушения (по геометрии) различают:
1. Сплошная или общая коррозия, ее условно подразделяют на:
а) равномерная, она протекает примерно с одинаковой скоростью по всей поверхности металла;
б) неравномерная – протекает с неодинаковой скоростью на различных участках поверхности металла;
в) структурно-избирательная коррозия, при которой разрушается преимущественно одна структурная составляющая сплава (обесцинкование латуни).
2. Местная коррозия, охватывающая лишь некоторые участки поверхности металла:
а) пятнами (латунь в морской воде);
б) язвами (коррозия стали в грунте);
в) точечная (питтинг) точек 0,1-2 мм (нержавеющая сталь в морской воде);
г) сквозная (сквозное разрушение металла);
д) подповерхностная начинается с поверхности, но распространяется под поверхностью (луженое железо);
е) межкристаллитная (интеркристаллитная) коррозия распространяется оп границам кристаллов. Этот вид особенно опасен, т.к. ведет к быстрой потере прочности и пластичности.
По механизму протекания коррозионного процесса как ОВР, зависящему от характера внешней среды различают:
а) химическую коррозию;
б) электрохимическую коррозию.
Скорость коррозии зависит от: характера среды, состояния поверхности, температуры и других факторов.
§ 1. Химическая коррозия.При химической коррозии металл разрушается за счет химической реакции в средах, не проводящих электрический ток. Это коррозия в газах и парах при высокой температуре в отсутствии влаги (газовая коррозия) или в жидкостях, не проводящих электрического тока (в неэлектролитах). Химическая коррозия возможна, если DG<0.
Химическую коррозию в атмосфере кислорода в общем виде можно представить как:
nMe +  O2 = MenOm (15.1)
O2 = MenOm (15.1)
Скорость окисления металла зависит от скорости собственно химической реакции и скорости диффузии окислителя через окисную пленку. Защитное действие пленки тем выше, чем лучше ее сплошность и ниже диффузионная способность.
Сплошность пленки на поверхности металла оценивается фактором Пиллинга-Бедвордса – VOX /VМе, где VOX – объем образовавшегося оксида; VМе – объем металла, израсходованного на образование этого оксида.
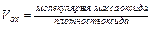 (15.2)
(15.2)
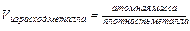 (15.3)
(15.3)
Если VOX /VМе<1, пленка не может быть сплошной и защищать металл от коррозии. Такая закономерность наблюдается для щелочных и щелочноземельных металлов. Отмечается линейный закон роста пленки:
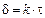 , (15.4)
, (15.4)
где d – толщина пленки; k – константа; t – время образования окисной пленки.
Если VOX /VМе>1, то пленка сплошная, коррозия будет тормозиться диффузией реагентов через пленку и по мере утолщения оксида рост ее будет замедляться. В этом случае рост пленки соответствует параболическому закону
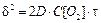 , (15.5)
, (15.5)
где D – коэффициент диффузии; С[О2] – концентрация кислорода.
Характерно для таких металлов как Fe, Co, Ni, Mn, Ti.
Для таких металлов как Al, Zn, Cr установлена логарифмическая зависимость роста пленки во времени (12.5) и VOX/VМе>1 пленки тонкие, сплошные.
δ = k ln τ (15.6)
В зависимости от температуры для одного и того же окисляющегося металла могут проявляться различные законы роста оксидной пленки.
При химической коррозии окисление и восстановление происходят одновременно, в одну стадию без «территориального» разделения.
§2. Электрохимическая коррозия подчиняется законам электрохимической кинетики гетерогенных реакций. Она протекает в средах, проводящих электрический ток: во влажной атмосфере с растворенными в ней газами, в растворах электролитов; в почве и расплавах солей. Термодинамическая возможность электрохимической коррозии с кислородной и водородной деполяризацией определяется прежде всего электрохимической диаграммой воды и наличием гальванической пары элементов (г.э.).
Согласно теории микрогальванопар (Де ля Рив), причиной электрохимической коррозии металлов является наличие на их поверхности микроскопических короткозамкнутых гальванических элементов, возникающих вследствие неоднородности металла и его контакта с окружающей средой. В отличие от специально изготовляемых в технике г.э. они возникают на поверхности металла самопроизвольно.
Электрохимическое растворение металла состоит из трех основных стадий:
1) анодного процесса – окисление Ме – nе– = Меn+ – перехода ионов коррозирующего металла в раствор под влиянием гидратации (сольватации) с освобождением электронов;
2) омического процесса – перетекания освободившихся электронов от анодных участков к катодным (поляризация);
3) катодного процесса – ассимиляции освободившихся электронов каким-либо деполяризатором, находящимся в растворе.
На анодном участке, потенциал которого электроотрицательнее, происходит растворение металла:
Men+×n+ mH2O = Men+×mH2O + n
| электронейтральный атом на поверхности анода | гидратиро-ванный ион в растворе | освободившиеся электроны на аноде |
Часть освободившихся электронов перейдет с анода на катод. Происходит поляризация электродов в результате чего jК и jА сближаются, что препятствует протеканию коррозионного процесса. Однако в растворе всегда имеются ионы или вещества (деполяризаторы), которые способны отводить электроны с катодных участков. Процесс отвода электронов, с катодных участков называется деполяризацией. Следовательно, электрохимическая коррозия может протекать, если электроны с анодных участков постоянно перетекают на катодный, а с катодного удаляются деполяризаторами.
Очень часто электрохимическая коррозия возникает в результате различной аэрации, т.е. неодинакового доступа кислорода воздуха к отдельным участкам поверхности металла. При коррозии железа под каплей электролита около ее краев, куда кислороду легче проникнуть будет катодный участок, а в центре капли, где толщина капли больше и О2 проникнуть труднее – анодный участок – происходит разрушение металла.
Коррозия железа проходит в две стадии: Fe0 ® Fe2+ ® Fe3+
Fe0 + 2H2O ® Fe(OH)2 ¯+ H2 осадок грязно-зеленого цвета
4Fe(OH)2 + 2H2O + O2 ® 4Fe(OH)3 ¯ осадок бурый (ржавчина).
Причем
А.: 2Fe0 – 4® 2Fe2+ К.: 2H2O + O2 + 4® 4OH–,
Тогда: Fe2++2OH–®Fe(OH)2.
Электрохимическая коррозия особенно опасна при контакте металлов с резко отличающимися значениями электродных потенциалов. Скорость коррозии пропорциональна ЭДС = jOX – jMe. Чем больше ЭДС, тем больше напряжение и ток, тем больше масса металла будет корродировать согласно закону Фарадея.
Например, коррозия латуни (сплав Zn и Cu) в растворе HCl.
 = –0,76 В,
= –0,76 В,  = +0,34 В
= +0,34 В
ЭДС = +0,34–(–0,76) = 1,1 В.
Образуется короткозамкнутый гальванический элемент Zn½HCl½Cu
А: Zn – 2® Zn2+; K: 2H+ + 2® H2.
Zn0 + 2H+ = Zn2+ + H20 – токообразующая реакция.
§ 3. Защита от коррозии. Причины, вызывающие коррозионные разрушения, многочисленны. Соответственно, разнообразны и методы защиты. Основная схема электрохимической коррозии диктует и методы защиты:
1) обработка внешней (коррозионной) среды;
2) защитные покрытия;
3) электрохимическая защита;
4) изготовление специальных коррозионно-устойчивых сплавов (легирование);
5) рациональное конструирование.
Методы защиты нужно рассматривать, исходя из основной схемы электрохимической коррозии (приведена в «Практикуме» стр.71).
1. Сущность метода «обработка внешней среды» заключается в удалении или понижении активности веществ, способствующих коррозии: а) нейтрализации кислых сред, вызывающих коррозию за счет водородной деполяризации; б) деаэрации – удаление О2 осуществляется кипячением воды, в) продуванием воды инертным газом, г) введением в воду восстановителей, д) добавлением ингибиторов коррозии в раствор.
В качестве ингибиторов в воде и водных растворах применяются некоторые окислители–пассиваторы: нитраты, хроматы, бихроматы, персульфат натрия и пленкообразователи (фосфаты, амины). Замедлители атмосферной коррозии подразделяются на нелетучие (контактные) и летучие (парофазные). Ингибиторы избирательно адсорбируются на анодных или катодных участках и блокируют эти участки, тем самым замедляя или прекращая коррозию. По механизму воздействия ингибиторы разделяются на анионные, катионные и неионогенные.
2. Защитные покрытия. Роль защитных покрытий сводится к изоляции металла от воздействия внешней среды. На поверхность металла наносятся:
а) металлические покрытия, они подразделяются на анодные и катодные. К катодным покрытиям относятся покрытия, потенциал которых в данной среде имеет более положительное значение, чем потенциал основного металла, например, луженое железо (рис. 15.1б): 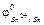 = –0,14 В,
= –0,14 В, 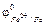 = –0,44 В.
= –0,44 В.
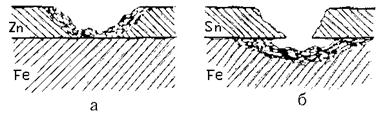
Рис. 15.1. Коррозия железа при нарушении металлических покрытий:
а – оцинкованное (анодное покрытие); б – луженое (катодное покрытие).
А: Fe – 2® Fe2+
К: 2H2O + O2 + 4®4OH–
Идет разрушение металла-изделия.
У анодного покрытия j покрывающего металла более отрицателен, чем основного (защищаемого) Ме (рис. 15.1а).
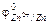 = –0,76 В;
= –0,76 В;  = –0,44 В.
= –0,44 В.
А.: Zn – 2® Zn0
К.: 2H2O + O2 + 4® 4OH–
Пока защитный слой полностью изолирует основной металл от окружающей среды, принципиального различия между катодными и анодными покрытиями нет. При нарушении целостности анодное покрытие разрушается, а изделие – нет.
Кроме того, к металлическим покрытиям относятся также покрытия, полученные металлизацией – на защищаемое изделие с помощью сжатого воздуха распыляется расплавленный другой металл. Возможность нанесения покрытия на собранные конструкции и хорошее сцепление с основой дает этот метод.
Термодиффузионные покрытия – этот способ широко используется для получения жаростойких покрытий (алитирование, хромирование, титанирование). Изделие помещают в смесь, содержащую порошок покрытия. При высоких температурах происходит диффузия наносимого металла в основной металл-изделие.
Плакирование – покрытие сплава чистым металлом. Толщина плакирующего слоя не превышает 5-10% от толщины сплава.
Воронение – нанесение на сталь оксидных пленок горячим раствором (NaOH + NaNO3) и образование пленки Fe3O4 (FeO, Fe2O3) цвета «воронова крыла».
Фосфатирование – образование фосфатной пленки на стали. Изделие при 95-98°С погружают в 3% раствор «мажефа» (Mn(H2PO4)2 и Fe(H2PO4)2) выдерживают в этом растворе до прекращения выделения Н2 с последующей кристаллизацией фосфатов Fe и Mn в виде черного осадка.
Анодирование – Al и Ti и других вентельных металлов с образованием оксидной пленки.
Органические покрытия: лаки, краски, эмали на основе органических полимеров; гуммирование – нанесение на металл сырой резины с последующей вулканизацией и образованием покрытия на металле.
3. Метод электрохимической защиты основан на торможении анодных или катодных реакций коррозионного процесса.
Уменьшение или полное прекращение коррозии достигается созданием на защищаемом изделии высокого электроотрицательного потенциала.
а) протекторная защита – защищаемое изделие соединяют с металлом, имеющим высокий электроотрицательный потенциал.
 . Протектор является анодом и растворяется, защищаемое изделие – катод. Например, корпус морского корабля (сталь) – протектор может быть Al, Mg, Zn.
. Протектор является анодом и растворяется, защищаемое изделие – катод. Например, корпус морского корабля (сталь) – протектор может быть Al, Mg, Zn.
А.: Mg – 2®Mg2+
К.: 2H2O + O2 + 4®4OH–
б) катодная защита – защищаемое изделие подключается к отрицательному полюсу внешнего источника тока, он становится катодом, а анодом служит вспомогательный металлический электрод. Анод растворяется, на защищаемом изделии (катоде) выделение Н2:
А.: Ме – n®Men+;
К.: 2H+ + 2®H2.
Коррозию подземных сооружений вызывают так называемые «блуждающие токи». Они обусловлены утечкой постоянного тока с рельсов электрического транспорта в землю. Место входа «блуждающего тока» в подземное металлическое сооружение является катодом, а место выхода из него – анодом. Для защиты от коррозии находят анодный участок методом дренажа и соединяют его с источником «блуждающего тока» (трамвайным рельсом), тогда весь ток проходит по этому проводнику и коррозия не происходит.
4. Изготовление специальных коррозионноустойчивых сплавов (легирование). Легирование – это введение при выплавке основного металла коррозионноустойчивых компонентов, вызывающих пассивацию металла. Сталь легируют: Cr, Mo, Ni, Co и другими металлами. Легирующие компоненты добавляются согласно правила Таммана – порог коррозионной устойчивости кратен 1/8 атомной доли легирующего компонента.
5. Рациональное конструирование. При конструировании важно предвидеть контактирующие детали металлического сооружения, которые могут подвергнуться интенсивной коррозии. В многоэлектродном элементе разделение на катод и анод не столь однозначно, как у двухэлектродных систем и полярность электродов может изменяться в зависимости от условий. Важно отметить, что железо, как основной металл, подвергающийся коррозии, необходимо «сделать» катодом.
Итак, коррозию металла можно затормозить или даже прекратить. Выбор способа защиты определяется его эффективностью, а также экономической целесообразностью.
Лекция 16
КООРДИНАЦИОННЫЕ СОЕДИНЕНИЯ
В данной лекции мы рассмотрим третий тип основных химических реакций – реакций комплексообразования, реализуемых за счет неподеленных пар электронов (орбиталей) молекул, ионов и вакантных (пустых) орбиталей ионов металлов. Отметим, что это преимущественно – переходные металлы, имеющие вакантные d-орбитали. Ранее мы рассмотрели с вами реакции кислотно-основного равновесия (с участием и переносом протона) и окислительно-восстановительные (с участием и переносом электрона). Молекулы и ионы с неподеленными парами называют лигандами, которые координируются вокруг центрального иона, имеющего вакантные орбитали с определенной степенью насыщенности и направленностью химических связей.
Комплексные (координационные) соединения чрезвычайно широко распространены в живой и неживой природе, применяются в промышленности, сельском хозяйстве, науке, медицине. Так, хлорофилл – это комплексное соединение магния с порфиринами, гемоглобин содержит комплекс железа (II) с порфириновыми циклами (лигандами). Многочисленные минералы, как правило, представляют собой координационные соединения металлов. Значительное число лекарственных препаратов содержит комплексы металлов в качестве фармакологически активных веществ, например инсулин (комплекс цинка), витамин В12 (комплекс кобальта), платинол (комплекс платины) и т.д. В широком смысле слова почти все соединения металлов можно считать комплексными соединениями, а в растворах это – основная форма существования соединений металлов. Основателем координационной теории комплексных соединений является швейцарский химик Альфред Вернер (1866-1919); за работы в этой области ему в 1913 г. была присуждена Нобелевская премия по химии.
§ 1. Основная характеристика комплексных соединений. Комплексные соединения образуют как металлы, так и неметаллы. В дальнейшем мы будем рассматривать в основном комплексные (координационные) соединения металлов. Комплексное соединение (сокращенно – комплекс) состоит из центрального атома (иона) металла-комплексообразователя М (здесь не указан его заряд), с которым связаны лиганды – L (старое название – адденды). Атом М и лиганды L образуют внутреннюю сферу комплекса (или внутреннюю координационную сферу). Внутренняя координационная сфера обычно при написании формулы соединения заключается в квадратные скобки. Лигандами могут быть нейтральные молекулы (обычно основного характера), отрицательно заряженные анионы (ацидогруппы). Простые положительно заряженные катионы в роли лигандов не выступают. Если внутренняя сфера комплекса несет отрицательный или положительный заряд, то для компенсации этого заряда (поскольку все индивидуальные соединения электронейтральны) необходимы ионы, образующие внешнюю
сферу. Во внешней сфере могут находиться не только ионы, но и нейтральные молекулы, очень часто – молекулы воды. Приведем несколько примеров:
1. Рассмотрим комплекс состава K4[Fe(CN)6]×ЗН2О – тригидрат гексацианоферрата (II) калия (ферроцианид калия, желтая кровяная соль). Здесь в роли центрального атома металла-комплексообразователя выступает ион железа (II), в роли лигандов – шесть одинаковых цианогрупп (ионов) CN–. Вместе атом железа (II) и шесть цианогрупп образуют внутреннюю координационную сферу комплекса, что в написанной выше химической формуле соединения обозначено квадратными скобками. Во внешней сфере в данном случае находятся четыре катиона калия (они компенсируют отрицательный заряд внутренней сферы) и три молекулы воды.
2. Рассмотрим теперь комплексы платины (II) состава транс-[Рt(NН3)2Сl2], [Pt(NН3)4]Cl2 и K2[PtCl4]. В первом комплексе внешняя сфера отсутствует, поскольку внутренняя координационная сфера электронейтральна. У второго комплекса во внешней сфере имеются два хлорид-иона (Сl–), так как внутренняя сфера комплекса [Pt(NН3)4]2+ несет положительный заряд, равный +2. В третьем комплексе во внешней сфере находятся два катиона калия (К+), поскольку внутренняя сфера [PtCl4]2– несет отрицательный заряд. Иногда в роли внешней сферы одного комплекса выступает внутренняя сфера другого комплекса, например, как в соединениях состава:
[Pt(NH3)4][PtCl4], [Со(NH3)4][Сг(СN)6], [Co(NH3)5NO2][Co(NH3)2(N02)4]2.
Нейтральные молекулы (но не ионы!), находящиеся во внешней сфере, называют (за исключением молекул воды или другого растворителя) клатратными молекулами, а сами такие соединения – клатратными соединениями (соединениями-включения). Например в комплексе никеля (II) состава NiI2·10А, где А – молекула карбамида (мочевины) – OC(NH2)2, на один атом никеля приходятся десять молекул карбамида, однако только шесть из них входят во внутреннюю сферу комплекса; четыре остальные молекулы карбамида и два иодид-иона (I–) образуют внешнюю сферу. Эти четыре молекулы карбамида являются клатратными молекулами. Состав комплекса в целом можно представить следующим образом: [NiA6]I2×4A.
Заметим, что иногда клатратами называют химические соединения, образуемые растворенными газами (Cl2, О2)в водных растворах.
Лиганд L образует с металлом-комплексообразователем М координационную связь различной химической природы (ионная, ковалентная, полярная; по происхождению – донорно-акцепторная, дативная или иной природы). Координационная связь может быть ординарной (одинарной), двойной. Координационные соединения, в которых два и более центральных иона, соединенных мостиковыми лигандами (m), называют полиядерными (многоцентровыми).
§ 2.Координационное число(к.ч.,или n) центрального атома М – это число координационных связей, образуемых атомом (ионом) металла-комплексо-образователя с лигандами. К.ч. может иметь значения от 2 до 12 (например, для некоторых соединений редкоземельных металлов). Наиболее часто встречаются к.ч. 4, 6. Ясно, что с точки зрения гибридизации первые – это тетраэдр или квадрат, вторые – октаэдр. Например, для n=4:
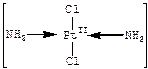

Число координационных связей, образуемых одним и тем же лигандом с одним атомом металла-комплексообразователя, называется дентатностью лиганда (старое название – координационная емкость). Лиганды могут быть монодентатными и полидентатными (би-, три-, тетра-, пента-, гексадентатными).
К монодентатным лигандам относятся F–, СI–, С, Br–, I–, H–, CN–, RNH2, NH3, H2O и т.д. Они образуют только одну координационную связь (если исключить из рассмотрения возможность образования мостиковых связей между двумя атомами металла М-CN-М).
| Бидентатные лиганды функционируют, например, в комплексах [Соеn2СО3] октаэдрического строения (en – сокращенное обозначение молекулы этилендиамина NH2-CH2-CH2-NH2): Здесь каждая из двух молекул этилендиамина реализует бидентатную координацию в металлоцикле: К тридентатным лигандам относится, например, триаминопропан NH2CH2-CHNH2-CH2NH2 в комплексе (к.ч. 6): | 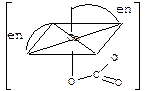
|
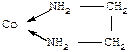
|
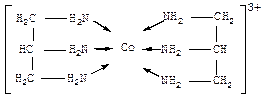
В роли гексадентатного лиганда может выступать, например, анион этилендиаминтетрауксусной кислоты:
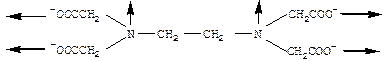
Полидентатные лиганды не обязательно должны реализовать свою макси-
мальную дентатность; они могут занимать и меньшее число координационных мест во внутренней сфере комплекса.
Иногда понятия координационного числа центрального атома М или дентатности лиганда формально становятся неопределенными, например, в ферроцене Fе(С5Н5)2 – ди-p-циклопентадиенилжелезе, в дибензолхроме Сr(С6Н6)2:
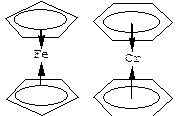
Ферроцен Дибензолхром
Здесь атом металла (железа или хрома) связан не с каким-то определенным донорным атомом лиганда, а целиком со всем лигандом – циклопентадиенилом (С5Н5), (однократно депротонированным остатком молекулы циклопентадиена) или молекулой бензола. Соединения подобного типа называют p-комплек-сами, или ароматическими комплексами металлов (если лиганд – ароматическое соединение; понятие «p-комплекс» – более широкое, чем понятие «ароматический комплекс»), еще их называют «сэндвичевыми».
Комплексные соединения могут быть катионного типа, анионного типа и комплексами-неэлектролитами.
Внутренняя сфера комплексов катионного типа несет положительный заряд: [Ag(NН3)2], [Cu(NH3)4]2+, |PtEn2]2+, [Co(NCS)2En2]+. Внутренняя сфера комплексов анионного типа несет отрицательный заряд: [Ag(S2O3)2]3–, [Sb(OH)6]–, [Co(NO2)6]3–, |HgI4]2–, [Pt(SCN)4]2–. Внутренняя сфера комплексов-неэлектроли-тов не несет никакого электрического заряда: [Pt(NH3)2Cl2], [Ni(ДМГ)2], где ДМГ-молекула диметилглиоксима (Н3С–С=NОН)2, [Рt(NН3)2СL4], [ZnL2], где L – молекула 8-оксихинолина.
Если комплекс содержит только один атом металла-комплексо-образователя, то он называется одноядерным (моноядерным); если он содержит два или более атомов центрального металла, то он называется многоядерным или полиядерным (биядерным, триядерным). Так, например, комплекс палладия (II) [PdCl4]2–, имеющий только один центральный атом – атом палладия (II), является одноядерным, а комплекс платины (II) [Pt2(NH3)2Cl2], содержащий два атома платины (II), – биядерным, [Pd6Cl14]2– – гексаядерным.
Если полиядерные комплексы содержат атомы металла одинаковой химической природы, то они называются гомометаллическими; если же в полиядерном комплексе имеются атомы металла-комплексообразователя разной химической природы, то такие комплексы называются гетерометаллическими. Так, из двух биядерных комплексов
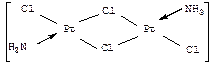
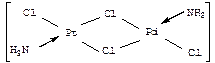
первый является гомометаллическим (оба центральных атома — атомы
платины (II), а второй — гетерометаллическим (содержит один атом платины и один атом палладия).
Если лиганд связан в полиядерном комплексе одновременно с двумя или даже с тремя атомами металла, то такой лиганд называется мостиковым лигандом, как, например, два m-хлоролиганда. В многоядерных комплексах могут осуществляться также связи металл-металл. Если связей металл–металл достаточно много (обычно – больше трех), то такие комплексы называют «кластерными соединениями» или просто «кластерами» (слово «кластер» в переводе на русский язык означает «рой», «скопление»).
Говоря о природе хим. связи, рассмотрим комплексный катион тетраамин меди ([Cu(NH3)4]2+), к.ч. 4 – 4 хим. связи. Каждая такая координационная связь Cu2+ – аммиак образуется за счет оттягивания «свободной» электронной пары от атома азота молекулы аммиака на пустую атомную орбиталь Cu2+, что в записи химической формулы обозначается стрелкой, направленной от донора к акцептору. Донором электронной пары, таким образом, является молекула аммиака, акцептором электронной пары – 4d-орбитали Cu2+. В результате обобществления электронных пар и образуются четыре координационные связи Cu2+ – аммиак, строение – квадратная конфигурация, в то же время для [Cu(NH3)6]2+ строение – октаэдр, обуславливающее интенсивно синий цвет.
В комплексном ферроцианид-ионе [Fe(CN)6]4– имеются шесть координационных связей железо (II) – цианогруппа.
Механизм образования таких связей в качественной форме можно представить следующим образом. Цианид-ион CN– имеет «свободную» электронную пару, которая оттягивается при комплексообразовании к атому Fe2+, в результате чего возникает s-донорно-акцепторная (от лиганда к металлу) составляющая координационной связи. С другой стороны, атом Fe2+ с d-электронной конфигурацией 3d6 может донировать свои d-электроны на пустую разрыхляющую молекулярную p-орбиталь цианогруппы, в результате чего возникает p-дативная (от металла к лиганду), составляющая координационной связи. В итоге обобществления электронов как лигандов (цианогрупп), так и металла-комплексообразователя (железа (II)) возникает двойная ковалентная полярная координационная связь железо (II) – цианогруппа. Вследствие всех этих перераспределений электронной плотности в шести координационных связях Fe2+ – цианогруппа положительный заряд на атоме Fe2+ уже не равен двум, а отрицательный заряд на цианогруппе также уже не равен минус единице. Другими словами, в действительности в ферроцианид-ионе [Fe(CN)6]4– нет ни катиона Fe2+, ни анионов CN–, а существует устойчивая группировка связанных между собой металла-комплексообразователя и лигандов.
Аналогичная картина характерна и для большинства других комплексов металлов, в которых многозарядные катионы металлов в действительности не существуют, поскольку комплекс состоит не из “чистых” ионов, а из центрального атома металла и лигандов, обобществивших свои электроны.
Отметим, что сказанное относится к частицам внутренней координационной сферы. Во внешней же сфере присутствуют катионы и анионы с зарядами, соответствующими их ионному состоянию. Например, в комплексе K4[Fe(CN)6] во внешней сфере имеются четыре катиона калия К+; во внешней сфере комплекса [Ag(NH3)2]Cl находится хлорид-анион Сl–.
Вышеприведенное краткое рассмотрение природы координационных связей в рамках метода валентных связей является упрощенным и лишь в качественной форме отражает реальную картину. В настоящее время наиболее распространенными являются три подхода к пониманию природы химической связи в координационных соединениях металлов: теория кристаллического поля, метод валентных связей и теория (метод) молекулярных орбиталей. Наиболее строгую картину можно получить с помощью метода молекулярных орбиталей. Геометрическая конфигурация строения внутренней сферы комплексов бывает различной: линейная, треугольная, квадратная, тетраэдрическая, октаэдрическая, пирамидальная, бипирамидальная и т.д., в зависимости от природы центрального атома металла, лигандов, внешнесферного окружения. Структура комплексов экспериментально обычно устанавливается рентгеноструктурным и спектральными методами. Важно отметить, что в комплексных соединениях d-орбитали, участвующие в образовании «дополнительных» химсвязей, расщепляются в соответствии с их геометрией и природой лигандов, так что образование комплекса приводит к дополнительному выигрышу энергии (устойчивость) по сравнению со свободными лигандами и ионом-комплексообразователем. Но это предмет рассмотрения теории поля лигандов в химии комплексных соединений.
Координационные соединения образуются, как правило, за счет донорно-акцепторной связи, то есть неподеленные пары электронов лигандов занимают вакантные места на орбиталях центрального атома. При этом количество неспаренных электронов и магнитный момент ионов-комплексообразователей остается таким же, как и у свободного иона в газовой фазе. Это справедливо, например, для аквакомплексов переходных металлов, например железа (II):
| 3d | 4s | 4p | 4d | |||||||||||
| Fe2+ | ¯ | | | | | |||||||||
| [Fe(H2O)6]2+ | ¯ | | | | | ¯ | ¯ | ¯ | ¯ | ¯ | ¯ | |||
| [Fe(CN)6]4– | ¯ | ¯ | ¯ | ¯ | ¯ | ¯ | ¯ | ¯ | ¯ |
Однако существуют также магнитно-аномальные комплексы, магнитный момент которых ниже, чем у газообразного иона. Их электронную структуру можно объяснить в рамках метода валентных связей следующим образом. Очень многие комплексные соединения имеют координационное число шесть. Шесть лигандов симметрично расположены в вершинах октаэдра. Для того чтобы получить шесть гибридных орбиталей, в их образовании должны принять участие шесть валентных орбиталей центрального атома: такое перераспределение электронной плотности называется sp3d2-гибридизация (сравните с sp3-гибридизацией атома углерода в алканах, где четыре связи направлены к вершинам тетраэдра). Обратите внимание, что в образовании гибридных орбиталей принимают участие d-орбитали с таким же порядковым номером, что и s-, p-орбитали. Это объясняется тем, что расположенные ниже по энергии внутренние d-орбитали заняты собственными электронами иона металла. Для того чтобы занять расположенные ниже по энергии орбитали, лиганды должны вынудить собственные электроны иона металла «спариться» и освободить внутренние d-орбитали для так называемой d2sp3-гибридизации. Это могут сделать только лиганды сильного поля, образующие прочные связи с ионом металла, например, цианид-ионы в комплексном гексацианоферрате (II) (см. выше).
Соответственно первый тип комплексов, обладающий высоким магнитным моментом, называются внешнеорбитальными комплексами, а второй тип с пониженным магнитным моментом – внутриорбитальными комплексами. Это различие, приводящее к изменению числа неспаренных электронов в комплексе, приводит к изменению магнитных моментов внешнеорбитальных и внутриорбитальных комплексов соответственно, вызвано энергетической неравноценностью соответствующих d-орбиталей (обычно ее называют энергией расщепления в поле лигандов и обозначают D или 10Dq).
По способности образовывать внутриорбитальные комплексы (по величине) все лиганды можно расположить в ряд, который называется спектрохимическим рядом лигандов.
CN–>NO2–>SO32–>NH3>NCS–>H2O>OH–>F–>Cl–>Br–>I–
Он получил свое название, потому что окраска комплекса зависит от положения лиганда в этом ряду, и в этом проявляется связь оптических и магнитных свойств координационных соединений.
Известно, что большинство важных на практике химических реакций протекают в растворах, к ним относятся также и реакции комплексообразования, поэтому в следующем разделе мы рассмотрим магнитные свойства растворов, в которых соединения переходных металлов реализуются в виде комплексов.
§ 3. Комплексные соединения в растворах. Комплексные соединения катионного и анионного типа чаще всего растворимы в воде; в их водных растворах устанавливаются химические равновесия, иногда довольно сложные. Комплексы-неэлектролиты, как правило, малорастворимы в воде; растворившаяся часть комплексов ведет себя как слабый электролит.
Так, при растворении аммиачного комплекса серебра (I) [Ag(NH3)2]Cl или ферроцианида калия K4[Fe(CN)6] вначале происходит первичная электролитическая диссоциация – отщепляются ионы внешней сферы:
[Ag(NH3)2]Cl ® [Ag(NH3)2]+ + Сl–, K4[Fe(CN)6] ® 4K+ + [Fe(CN)6]4–
При первичной диссоциации комплекса, имеющего ионы внешней сферы, соединение ведет себя как сильный электролит – полностью отщепляет ионы внешней сферы. Затем происходит вторичная диссоциация комплекса уже по типу слабого электролита – отщепляются лиганды внутренней сферы.
Так, в случае аммиачного комплекса серебра наблюдается последовательное замещение молекул аммиака молекулами воды:
[Ag(NH3)2]+ + Н2О = [Ag(NH3)Н2О]+ + NH3
[Ag(NH3)Н2О]+ + Н2О = [Ag(Н2О)2]+ + NH3
Каждой ступени диссоциации внутренней сферы комплекса соответствует состояние ступенчатого химического равновесия, характеризующееся своей константой химического равновесия. Заметим, что при написании химических уравнений процессов диссоциации внутренней сферы комплекса в водных растворах молекулы воды чаще всего для краткости не записываются. Например, вместо уравнения
[Cu(NH3)4]2+ + Н2O = [Сu(NH3)3H2O)]2+ + NH3
упрощенно записывают
[Сu(NН3)4]2+ = [Сu(NН3)4]2+ + NH3
Рассмотрим процесс образования комплекса в растворе в общем виде (без указания зарядов М и лигандов): М + nL = MLn.
Поскольку это уравнение отражает химическое равновесие, то можно записать выражение для константы равновесия b:
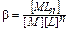 , (16.1)
, (16.1)
где все концентрации [MLn], [М] и [L] – равновесные. Величина b называется константой устойчивости комплекса. Обратная ей величина Кн, соответствующая равновесию диссоциации комплекса MLn = M + nL, выражаемая формулой:
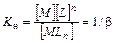 (16.2)
(16.2)
называется константой нестойкости или константой неустойчивости того же комплекса. Чем больше b, тем прочнее комплекс. Для устойчивых комплексов величина b имеет довольно высокие значения.
В качестве примера приведем константы устойчивости некоторых комплексов для водных растворов при температуре 20-30°С:
Cu2+ + 4NH3 = [Cu(NH3)4]2+ 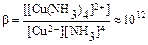
Al3+ + 4OН– = [Al(OH)4]– 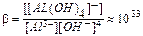
Знание констант устойчивости комплексов позволяет рассчитывать равновесные концентрации частиц в растворах, проводить сравнительную оценку прочности комплексов и т.д., что используется, например, в аналитической химии, при оптимизации технологических процессов, в медицине. Если, например, организм пересыщен соединениями какого-либо металла, что приводит к его отравлению различной степени сложности, то в организм вводят нетоксичные лиганды, которые образуют устойчивые растворимые комплексы с этим металлом, выводящиеся затем из организма. Если, напротив, в организме ощущается недостаток каких-либо металлов (например, дефицит железа при малокровии), то при лечении в организм вводят комплексные соединения этих металлов умеренной прочности.
Комплексные соединения участвуют в разнообразных химических реакциях, включая внутрисферное замещение одних лигандов на другие, диссоциацию лигандов, например молекул воды:
[PtCl3H2O]– + Н2О = [РtС13ОН|2– + Н3О+
Многие комплексные соединения являются катализаторами различных процессов гомогенного и гетерогенного катализа. Часто при получении лекарственных препаратов на основе фармакологически активных комплексных соединений удается понизить токсичность как металла, так и лигандов, связанных в комплексе, и модифицировать в желаемом направлении их биологическую активность.
§ 4. Номенклатура комплексных соединений. Комплексные соединения часто имеют довольно сложный состав и строение; к настоящему времени разработаны специальные системы составления их химического названия (номенклатуры). Существуют три основных подхода к номенклатуре комплексных соединений металлов.
1. Традиционные специфические вненоменклатурные названия, по которым различным комплексным соединениям присваивались те или иные наименования (например, K4[Fe(CN)6] – желтая кровяная соль, ферроцианид калия; К3(Fе(СN)6] – красная кровяная соль, феррицианид калия; [Pt(NH)4](OH)2 – первое основание Рейзе; транс–[Рt(ОН)2(NН3)2] – второе основание Рейзе; цис–[PtCl2(NH3)2] – хлорид Пейроне; К[Со(NO2)4(NН3)2] – соль Эрдмана и т.д.).
2. Номенклатура, предложенная основателем координационой теории швейцарским химиком Альфредом Вернером, принятая в несколько измененном виде его последователями.
3. Номенклатура, рекомендованная в 1960 г. комиссией по номенклатуре неорганических соединений Международного союза по чистой и прикладной химии (ИЮПАК).
В современной научной, учебной, медицинской литературе используются все три подхода, хотя можно отметить тенденции к преимущественному применению номенклатуры, рекомендованной ИЮПАК, как наиболее логически обоснованной. В русском языке эта номенклатура несколько модифицирована, адаптирована к правилам русского языка.
Согласно второй – комплексные катионы вначале называют отрицательно заряженные лиганды внутренней сферы с окончанием «о» (хлоро-, бромо-, нитро-, родано-). Если их число больше одного, то перед названиями лигандов добавляют числительные ди-, три-, тетра-, пента-, гекса- и т.д. Затем называют нейтральные лиганды, причем молекулу воды называют «акво», молекулу аммиака – «аммин». Если число нейтральных лигандов больше одного, то добавляют числительные ди-, три-, тетра- и т.д.
После нейтральных лигандов называют атом металла-комплексо-образователя и в последнюю очередь – внешнесферные анионы. При этом наименование атома металла составляют из корня его латинского названия с характерным окончанием, зависящим от степени окисления центрального атома металла-комплексообразователя. Окончания эти следующие:
| Степень окисления | I | II | III | IV | V | VI | VII | VIII |
| Окончание | а | о | н | у | ан | он | ин | ен |
Например (для наглядности здесь и далее указана степень (состояние) окисления центрального металла, хотя обычно это не делается): [PtII(NН3)4]С12 – тетрамминплатохлорид; [PtIV(NН3)4С12]Cl2 – дихлоротетрамминплатехлорид; [CoIII(NН3)5С1]SO4 – хлоропентамминкобальтисульфат.
Для комплексных анионов вначале называют отрицательно заряженные лиганды с соответствующими числительными (ди-, три-, тетра-, и т.д.). Затем называют нейтральные молекулы, после чего идет название металла с соответствующим окончанием, характеризующим его степень окисления, к которому добавляют еще суффикс «ат». В заключение в родительном падеже называют внешнесферные катионы. Например: K[AgI(CN)2] – дицианоаргентаат калия; K2[PtCl6]] – гексахлороплатеат калия; K4[FeII(CN)6] – гексацианоферроат калия; Na3[FeIII(CN)6] – гексацианоферриат натрия.
Для нейтральных комплексов названия строят аналогично предыдущему, за исключением того, что к наименованию металла-комплексообразователя не прибавляют никаких окончаний.
Например: [PtII(NH3)2Cl2] – дихлородиамминплатина; [CoIII(NН3)3(NО2)3] – тринитротриамминкобальт; [CrIII(NH3)3(SCN)3] – трироданотриамминхром.
Третью номенклатуру можно рассматривать как усовершенствованную и модернизированную номенклатуру А. Вернера. Лиганды называются так же, как и в вернеровской номенклатуре, т.е. отрицательно заряженные лиганды оканчиваются на «о», а нейтральные имеют обычные наименования.
Одноядерные комплексы. Вначале называют катион, затем – анион. При перечислении лигандов сначала называют отрицательно заряженные, затем – нейтральные с соответствующими числительными (ди-, три-, тетра- и т.д.). После этого называют атом металла-комплексообразователя, указывая после названия металла его степень окисления римскими цифрами в круглых скобках. Символ (0) используют для обозначения нулевой степени окисления. Если комплекс представляет собой анион, то к названию центрального атома добавляют суффикс «ат». Если лиганды представляют собой сложные многоатомные молекулы, то вместо числительных ди-, три-, тетра- и т.д. используют числительные бис-, трис-, тетракис-, пентакис-, гексакис-. Например
[Рt(NН3)4]Сl2 – тетрамминплатина (II) хлорид;
[Рt(NН3)4Cl2]Cl2 – дихлоротетрамминплатина (IV) хлорид;
[Co(NH3)5Cl]SO4 – гексамминкобальт (III) сульфат.
Если катион называют в родительном падеже (что в русском языке допускается), то наименования катиона и аниона пишут раздельно: [Co(NH3)6]Cl2 – гексамминкобальт (III) хлорид или гексамминкобальта (III) хлорид.
| бис(этилендиамин)кобальт(III)– m–имидогидроксобис(этилендиа мин)-кобальт(III)–ион. |
Полиядерные комплексы можно проклассифицировать на гетероядерные и гетеровалентные (смешанно-валентные); в гетероядерных два или более металлоцентра – разные металлы, в гетеровалентных – металлоцентры одного металла, но в разных степенях окисления. Для них характерно взаимодействие d-орбиталей, приводящее к изменению магнитных, оптических свойств, а также явлению переноса (миграции) электрона между металлоцентрами.
Координационная химия (химия комплексных соединений) длительное время считалась одним из разделов неорганической химии; объяснялось это тем, что большинство известных ранее координационных соединений содержало в качестве лигандов, как правило, типичные неорганические молекулы и ионы – аммиак, воду, нитро-, роданогруппу и т.п. Экспериментальные исследования неорганических комплексных соединений были начаты за несколько десятилетий до того, как стала бурно развиваться органическая химия. После создания А. Вернером координационной теории химия комплексных соединений стала постепенно, в течение десятилетий, превращаться в самостоятельный раздел химической науки. Ее успешное развитие связано с именами Трассера, Цейзе, Йергенсена, Грэма, Клауса, Бломстранда, школы А. Вернера, а в 20 в. – Л.А. Чугаева, И.И. Черняева, А.А. Гринберга, Чатта, Найхольма, Фишера, Бьеррума и многих других ученых различных стран. В наши дни координационная химия — интенсивно развивающаяся в различных направлениях наука, тесно переплетающаяся с другими областями химии. Достаточно отметить, что в нашем организме все ионы металлов находятся в форме комплексных соединений (гемоглобин!).
Лекция 17
Обзор химических свойств элементов
Наконец, в последней лекции мы с Вами обобщим свойства элементов с точки зрения электронного строения согласно периодическому закону и таблицы элементов Менделеева с одной стороны, и химических реакций (кислотно-основных, окислительно-восстановительных, комплексообразования), – с другой.
Среди многообразия химических соединений и их свойств рассмотрим некоторые общие закономерности и их проявления.
Ранее отмечалось, что по своей электронной конфигурации элементы классифицируются на s-, p-, d-, f-элементы, которые занимают в таблице Менделеева определенные периоды и группы.
Первый из них – водород, который согласно электронному строению существует в следующих формах: H+ (протон), H (атомарный, он реализуется при электролизе на катоде), H2 (молекулярный, согласно закону Авогадро) и H– (гидрид). Именно первая и последняя формы определяют его дуализм, он проявляет свойства щелочных металлов и галогенов (например CaH2). И тем не менее он обладает индивидуальными свойствами, присущими только ему. H+ – кислота по определению, окислительные свойства выражены слабо, только при электролизе может восстанавливаться до водорода. В комплексообразовании (несмотря на вакантную орбиталь – акцептор) – «не замечен», более того является конкурентом ионов-комплексообразователей (особенно переходных элементов), поэтому в кислых и щелочных средах, где концентрация протонов и гидроксил-ионов соизмерима с концентрацией центральных ионов-комплексо-образователей и лигандов, они реализуются в форме кислот и оснований, т.е. в кислых и щелочных средах комплексы практически не реализуются (следует отметить, что в щелочных средах реализуются гидроксокомплексы, например, [Al(OH)4]–).
Атомарный водород – один из самых сильных восстановителей (сравните с молекулярным). В виду малых размеров для водорода характерно явление «наводораживания», особенно металлов, так один из лидеров, поглощающих этот газ – титан: на один объем поглощает до 800 объемов водорода, образуя титановую губку, который может иметь и полезное применение, например в топливных элементах. Следуя вниз по первой группе таблицы Менделеева, мы обнаружим электронные аналоги ns1(n–главное квантовое число и номер периода) – щелочные металлы, отдав один электрон образует устойчивую оболочку инертного газа. Они – электроположительны, потенциал восстановления – отрицательный, поэтому основная форма их существования – катионная (Na+,K+). И с точки зрения кислотно-основных свойств они образуют основания – щелочи (NaOH, KOH). Окислительные и комплексообразующие свойства выражены в водных растворах слабо.
Аналогично ведут себя с точки зрения кислотно-основных и окислительно-восстановительных свойств – щелочно–земельные металлы ns2. Следует отметить, что основные свойства у них слабее, чем у первых, а бериллий проявляет амфотерные свойства. С некоторыми лигандами – комплексонами они образуют комплексы, например кальций с трилоном–Б, что используется для «снятия» жесткости воды.
Коснемся правой колонки таблицы Менделеева, где расположены инертные газы с электронной конфигурацией ns2p6 (устойчивая оболочка), несклонные принимать и отдавать электроны, как известно они химически инертны, они одноатомны. Но есть и исключения, например ксенон образует с кислородом и фтором молекулярные соединения, в экстремальных условиях, что объясняется наличием пустых d-орбиталей у инертных газов и подходящим потенциалом ионизации кислорода и фтора. Рассмотрите и объясните их применение, например, в водолазной технике, светооптике и др.
Теперь целесообразно рассмотреть химию 2p-элементов (B, C, N, O, F), сразу отметим, что у них нет свободных d-орбиталей, а заполнение электронами p-орбиталей следует правилу Гунда и принципа Паули. Именно поэтому они обладают набором индивидуальных свойств, хотя и «открывают» соответствующие группы элементов – электронных аналогов, имеющих близкие химические свойства.
И первый из них бор, имеющий конфигурацию 2s2p1 , которую трудно отнести к типичным металлам или неметаллам. В природе он существует в основной своей форме В3+. Основными его минералами являются бораты: Na2B4O7×10H2O – бура, Na2B4O7×4H2O – кернит, H3BO3 – сассолин. Борная кислота – H3BO3 известна как слабая (Кдис=7,1×10–10). Формально отрицательная степень окисления его проявляется в металлических соединениях бора, твердых, жаростойких, химически устойчивых. В3+ образует массу соединений с более электроотрицательными элементами. Для него характерны бораты и бораны состава BxOy, BxHy со структурными фрагментами B-O-B, B-H-B с электроннодифицитными химическими связями (напомним, что в методе МО допускаются «дробные» связи с ковалентностью 0,5, 1,5). Для бора известны диборан B2H6 и боразол B2N3H6. Рассмотрите их структуры.
Химия углерода – это как известно химия органических и высокомолекулярных соединений, богатой на многообразие и многочисленность. Мы рассмотрим лишь неорганическую химию С с конфигурацией 2s22p2и возбужденной конфигурацией 2s1p3. С точки зрения гибридизации все рассматриваемые р-элементы – тетраэдры.
Что касается углерода, то он легко реализует С-С цепочки вершинами тетраэдров, ребрами – С=С и гранями – СºС, а также С-Н связи, как ковалентные, так и донорно-акцепторные. В нулевой степени окисления он встречается в виде аллотропных модификаций – алмаз, графит, и синтетических – карбин и фуллурен. В природе он «проявляется» преимущественно в степени окисления +4: нефть, уголь и карбонаты (СО32–). Степень окисления +2 известна для угарного газа (СО), «знаменитого» в доменных процессах (восстановитель!) и в экологии (ядовит!), как продукт неполного сгорания. Отрицательная степень окисления известна для карбидов металлов (карбид кальция непременный участник сварочных аппаратов). Основная степень окисления +4 по кислотно-основным свойствам известна для угольной и уксусной кислот. Окислительно-восстановительные и комплексообразующие свойства не характерны.
Переходим к азоту 2s2 2p3, для которого характерен больший набор электронных состояний от –3 до +5, от аммиака (гидразина) до азотной кислоты, от основных до кислотных свойств, от восстановительных до окислительных (HNO3 – и кислота, и окислитель). Более того азот, а в равной степени и кислород в низких степенях окисления обладают неподеленными парами и входя в состав органических соединений (-NH2, -СООН являются прекрасными лигандами – донорами неподеленных пар. Рассмотрите химию азота по более «толстым» учебникам.
Для кислорода (поистине элемента жизни) характерны формы: О, О2, О3 (экология) и многочисленные соединения со степенью окисления –2. Следует отметить и пероксогруппу [-O-O-]2–, разберите ее кислотно-основные и окислительно-восстановительные свойства и их применение в быту и технике. Кислород в нулевой степени окисления – окислитель.
Наконец перейдем к предпоследнему представителю этого периода фтору с конфигурацией 2s2p5. Он самый электроотрицательный и образует соединения со всеми элементами. Его высокая реакционная способность проявляется не только в OF2, но и в соединениях с инертными газами (ХеFх) его основная степень окисления –1, фториды металлов (фторид-ион «растворяет» стекло), что роднит его с галогенами. Для всех них типичны соли, галогениды металлов (NaCl, NaF, KBr, CuCl2 и т.д).
Рассматривая химию элементов 3–7 группы (сверху вниз) следует отметить, что p-элементы 3 периода являются как бы переходными в группах между 2 и 4периодами, т.е. химия кремния отличается от химии углерода (сверху) и химии германия, олова, свинца (снизу) при наличии общих свойств.
Так в подгруппе алюминия (алюминий, галлий, индий, таллий) характерная степень окисления +3 (у таллия есть соединения со с.о. +1). По кислотно-основным свойствам они амфотерны. Восстановительные свойства алюминия известны по реакции алюмотермии:
2Al+Fe2O3=2Fe+Al2O3
Для алюминия характерна способность образовывать в водных растворах с Н2О и ОН– полиядерные образования [Alx(OH)y(H2O)z]n+.
Наличие вакантных d-орбиталей позволяет ему образовывать комплексные соединения, простейшее – гидрат [Al(H2O)6]3+.
Наличие вакантных d-орбиталей, начиная с элементов третьего периода, позволяет им участвовать в реакциях комплексообразования.
Так, например, для подгруппы кремния характерны соединения типа [SiF6]2– с sp3d2 гибридизации октаэдрического строения, а для углерода это невозможно. Особенностью элементов главной подгруппы 4 группы, является заметно выраженное усиление металлических свойств в ряду углерод-свинец, а стало быть, основных свойств. Для олова и свинца характерны с.о. +2, +4, причем для олова (+2) она неустойчива в водных растворах (соединения олова (2) легко окисляются до олова (4), для них характерны амфотерные свойства).
Здесь следует отметить, что для ионов элементов у которых с.о. более 3+, характерны оксосоединения в водных растворах (Э=О), например, SnO2+, SO42–, MnO4–, VO2+ («аномальный» гидролиз воды с отщеплением двух протонов от кислорода молекулы воды).
Аналогично мы можем проанализировать свойства элементов подгруппы фосфора, для которых характерны соединения со с.о. –3 до +5, за исключением висмута, для которого характерны металлические свойства и с.о. +3. Здесь следует отметить специфику кислотно–основных свойств фосфора в высоких степенях окисления – многообразие фосфорных кислот. К слову сказать усиливающееся у серы. Для этих кислот характерны следующие структурные фрагменты: Э=О, Э-О-Э, S=S, Р-Н, Э-О-О-Э и др., способствующие их многообразию. Для серы характерны мостиковые структуры -S-S-S-, что используется при вулканизации резины, да и сама она существует в виде циклов S4, S6 (аморфные и кристаллические модификации серы).
Для соединений серы характерны со с.о. –2 до +6 (H2S, H2SO4), от восстановителя до окислителя.
Также для соединений подгруппы хлора характерны состояния со с.о. +1, +3, +5, +7 (напомним, что у фтора это не реализуется).
Следует отметить, что для соединений с промежуточными с.о. характерны реакции диспропорционирования (самоокисления-самовосстановления), например:
2Pb2+=Pb4++Pb 2Cl5+=Cl7++Cl3+ 2Cu+=Cu2++Cu
Все элементы побочных подгрупп d- и f-элементы – переходные металлы, для которых характерны реакции комплексообразования. Следует отметить, что f-элементы существуют в с.о. +3, редко +2 и +4 и мало различаются в хим. свойствах, поэтому и помещены в таблице Менделеева в одну «клетку». Конечно существует классификации элементов на металлы и неметаллы и т.д. Но это уже предмет неорганической химии, изучающей химию элементов и их соединений во всем многообразии.
Дополнение к лекции 12
О СТРУКТУРЕ ВОДЫ И РАСТВОРОВ
Значительно сложнее дело обстоит со структурой жидкой воды. В настоящее время в основе лежит представление о том, что в жидкой воде существует три… [Н2О]900–1000=[Н2О]4–5=[Н2О]1–2, которое смещается с повышением температуры в сторону образования пара и с понижением температуры в сторону образования…Учеб. пособие для вузов.
Е издание, исправленное и дополненное
Оригинал макет: Д.В. Фролов– Конец работы –
Используемые теги: курс, лекций, общей, химии0.061
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Курс лекций по общей химии
Что будем делать с полученным материалом:
Если этот материал оказался полезным для Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.196 сек.
Новости и инфо для студентов