рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Философия
- /
- ОТБОР ПРОБ С ВОДНЫХ ОБЪЕКТОВ
Реферат Курсовая Конспект
ОТБОР ПРОБ С ВОДНЫХ ОБЪЕКТОВ
ОТБОР ПРОБ С ВОДНЫХ ОБЪЕКТОВ - раздел Философия, Конспект лекций по курсу: Методы и способы измерений параметров окружающей среды ВСТУПЛЕНИЕ Речки И Ручейки. Пробы Воды Отбирают ...
Речки и ручейки. Пробы воды отбирают в местах наиболее быстрого течения в фарватере. Если не обусловлены какие либо особенные цели исследования, то не следует отбирать пробы из стоячей воды перед дамбой или сразу после нее в глухих рукавах. Дело в том, что химических состав воды в таких местах может значительно отличаться от среднего состава, который характеризует данный водный объект. В случае смешения вод двух рек или речной воды со сточной пробы для анализа необходимо отбирать в местах полного перемешивания водных масс, которые устанавливают специальными исследованиями. Пробы отбирают под поверхностью воды, лучше в верхней трети общей глубины, как правило на глубине 20-30 см.
Водохранилища, озера, пруды. Пробы воды отбирают на стационарных точках, размещенных по акватории, и , как правило, на двух глубинах – возле поверхности (0,2 – 0,5 м) и возле дна (0,5 м). На промежуточных глубинах пробы отбирают в зависимости от термической стратификации и при спец. исследованиях.
Смешанную пробу в водохранилищах, озерах и прудах отбирать не рекомендуется. В стоячих водах вследствие большой разницы между химическим составом проб в разных местах отдельные компоненты при смешивании воды могут взаимодействовать. В результате качество воды не будет отвечать химическому составу отдельных проб, которой был характерным до их смешения.
Атмосферные осадки (дождь, снег) и лед. Химический состав атмосферных осадков формируется в основном в воздухе и после их выпадения на поверхность земли осадки превращаются в разновидность природных вод.
Пробу дождевой воды улавливают широкой лейкой, трубка которой достигает дна посуды для пробы. Если необходимо определить средний химический состав дождевой воды, то ее отбирают на протяжении всего времени выпадения дождя. При необходимости определить качество чистой дождевой воды пробу отбирают через 5 – 10 мин. после начала дождя.
Снег улавливают в лейку или в широкую или глубокую чашку и отморораживают. Снежное покрытие отбирают в тех местах, где его шар самый толстый. В начале лопатой снимают верхний шар, который отбрасывают, а затем заполняют снегом широкогорлую посуду.
Лед отбирают кусками с различных мест очищают их поверхность долотом или ножом, кладут в чашку и выдерживают некоторое время, а затем переносят в посуду с широким горлом, в котором отмороживают при комнатной температуре. Мелкие кусочки льда засыпают в лейку Бюхтера, смывают горячей дистиллированной водой и переносят в посуду для пробы.
Консервация, транспортировка и хранение проб воды
Консервация проб воды имеет целью сберечь их физические свойства и химический состав в таком состоянии, в каком они были в момент отбора пробы. Консервацию проводят в тех случаях, когда нет возможности выполнить анализ на месте отбора пробы. Однако, необходимо помнить, что консервация проб воды не может полностью предотвратить изменение их химического состава, которое обусловлено протеканием в воде разнообразных физико-химических и биологических процессов. В связи с этим определение физических свойств и химического состава законсервированных проб желательно проводить на следующий день, но не позже, чем на третий день после отбора пробы воды.
Универсальность метода консервации природных вод нет, поэтому часто для определения различных ингардиетов отбирают отдельные пробы воды и по-разному их консервируют.
Пробы воды не консервируют или их нельзя консервировать при определении многих показателей: температуры, Eh, СО2, НСО3-, кислотности, щелочности, сульфидов, озона, хлора(сразу); растворенного кислорода (фиксируется сразу); вкуса, запаха, цветности (2 часа); биохимического употребления кислорода (сутки при 3-40 С); взвесей, мутности, прозрачности, удельной электропроводности, растворимых веществ, рН, ароматических углеводов, жирных кислот (сутки), фенолов (5 суток); Na+, K+, Ca2+, Mg2+, Cl-, SO42-, F-, оборотов (до 3 суток).
Наиболее распространенными консервантами воды являются следующие:
Ø 1 мл концентрированной H2SO4 на 1 л води при определении химического употребления кислорода (ХУК), Сорг, Nобщ, Nорг, NH4+, NO2-, NO3-, Zn2+;
Ø 5 мл концентрированной HNO3 на 1 л воды при определении ионов Mn2+, Cu2+, Ni2+, Cd2+, Pb2+, Ag+, Cr (III, VI);
Ø 2-4 мл хлороформа на 1 л воды при определении цветности, NH4+, NO2-, NO3, SO42-, фосфатов и синтетических ПАВ.
Иногда применяют специальные способы консервации, которые связаны с химическими свойствами определяемого ингардиента. Например, для определения общего содержания Fe пробу консервируют, добавляют 25 мл концентрированной HNO3 на 1 л воды. При этом гидроксиды и др. малорастворимые соединения Fe (II,III), а также их комплексы с органическими веществами природных вод разлагаются и все формы Fe переходят в аква-ионы Fe3+, которые определяют фотометрически. Если необходимо отдельно определить содержание железа (II) и железа (III), то пробу воды консервируют смесью уксусной кислоты с уксуснокислым натрием и хранят, не допуская контакта с воздухом.
Для определения растворённых сульфидов пробу консервируют, добавляя к 1 л. воды 10 мл. 10 % раствора ацетата Cd или Zn. При этом оседают малорастворимые сульфиды этих металлов, которые длительное время хранятся и могут быть потом определены после их растворения в серной кислоте.
Способы консервации проб воды.
Для транспортировки пробы воды отбирают в стеклянную или полиэтиленовую посуду с пробками и герметично закрывают. Необходимо следить за тем, чтобы под пробкой не оставались пузырьки воздуха. Пробы воды желательно хранить в холодильнике при температуре 3 – 4 0С. Анализировать пробы начинают после того, когда их температура сравняется с комнатной.
Определение химических инградиентов в растворённом состоянии, коллоидно-дисперсной форме и взвесях
В природных и сточных водах всегда содержатся растворенные и нерастворенные вещества. Поэтому определение концентрации многих химических инградиентов в нефильтрованной и фильтрованной воде дает разные результаты. Это особенно касается соединений ионов металлов, потому что они в значительной степени сорбируются на взвесях и коллоидных частицах. Поэтому часто возникает необходимость определить не только общее содержание определённых инградиентов, а также отдельно в взвесях, в коллоидном и в растворённом в воде состоянии. Например, установлено, что токсичность воды р. Дунай, которая является сильно мутной, почти полностью исчезает, если воду профильтровать. Это обусловлено сорбцией содержаний токсичных металлов, таких как Hg, Cd, Pb, Cr и др. на взвесях и коллоидных частицах.
Для выделения взвесей из природных вод применяют отстаивание проб воды, фильтрацию, центрифугирование и ультрафильтрацию (мембранная фильтрация). Наиболее распространены два последних метода. Фильтрация через мембранные фильтры с диаметром пор 300 – 500 нм (0,3 – 0,5 мк) или центрифугирование в течение 20 – 30 мин. при 7 – 8 тыс. об/мин. обеспечивает практически полное выделение взвесей.
Что касается выделения коллоидов, которые также являются хорошими сорбентами, нет единой точки зрения. Некоторые исследователи относят к коллоидам частицы размером 1 – 200 нм, другие – частицы, которые задерживаются на ультрафильтрах с диаметром пор 20 – 25 или 4 – 17 нм. В то же время установлено, что через целлофановые мембраны не диффундирует до 90 – 95 % высокомолекулярных закрашенных органических веществ природных вод. Однако их нельзя отнести к коллоидам, потому что они имеют главные признаки истинно растворённых веществ, а именно: термодинамически стабильные, в системе отсутствует поверхность раздела фаз, они являются гидрофильными соединениями и не выделяются даже при длительном центрифугировании.
Однозначное решение этой проблемы невозможно через то, что коллоидные свойства диспергированных в водной среде частичек обусловлены не только размерами, но также их структурой и химическими свойствами, которые влияют на их гидрофильность. Поэтому рациональным является такой подход, который объединяет 2 характеристики коллоидных и истинно растворённых высокомолекулярных веществ – их размеры и молекулярная масса.
Согласно этого подхода до взвесей относят частицы, размер которых превышает 300 нм. Их можно выделить фильтрацией природной воды через мембранный фильтр с диаметром пор 300 – 500 нм. В гидрохимической практике для этого рекомендуют использовать ультрафильтры с диаметром пор 450 нм.
К коллоидным отнесены диспергированные в воде частицы, размеры которых колеблются в пределах 5 – 500 нм. Очевидно, что взвеси и коллоиды можно выделить вместе фильтрованием пробы воды через мембранный фильтр с диаметром пор 5 – 10 нм. При этом через фильтр не пройдут также растворённые соединения с молекулярной массой 100 тыс. и больше.
Наконец, истинно растворённые соединения можно характеризовать их молекулярной массой до 300 тыс. или размерами, что не превышают 5 – 15 нм.
Содержание определённого химического инградиента в взвесях, в коллоидной форме и в истинно растворённом в воде состоянии определяют так. Вначале пробу воды обрабатывают кислотой или другим реагентом для перевода всех форм определяемого инградиента в истинно растворённое состояние и определяют его общую концентрацию С общ. Потом другую порцию этой же воды фильтруют через мембранный фильтр с размером пор 450 нм и в фильтрате определяют концентрацию С1. Наконец, третью порцию воды фильтруют через мембранный фильтр с размером пор 10 нм и в фильтрате определяют С2. Концентрацию определяемого инградиента в каждой форме определяют по формулам:
Свзвес = Собщ. – С1; Сколоид. = С1 – С2; Сраств. = С2.
Концентрирование микрокомпонентов
Концентрирование микропримесей неорганических и органических соединений целесообразно проводить после предварительной фильтрации воды или центрифугирования, для того, чтобы избавиться от нерастворённых соединений.
Для концентрирования микрокомпонентов используют 2 группы методов.
К первой из них принадлежит выпаривание и вымораживание, которые приводят к уменьшению общего объема пробы воды и, как следствие, к увеличению концентрации всех инградиентов.
Общий недостаток этих методов заключается в том, что при значительной концентрации главных ионов они выпадают в осадок в виде солей, например, СаСО3, СаSO4 и т.д. При этом на поверхности осадка способны сорбироваться органические и неорганические микропримеси, вследствие чего химический состав и содержание растворённых компонентов изменяется.
При вываривании пробы воды из неё могут выделяться не только осадки солевых компонентов, но и отгоняться летучие органические соединения. Установлено, например, что в период цветения днепровской воды, когда она насыщена органическими соединениями, результаты определения химического употребления О2 в кипяченной в течение 15 – 20 мин. воды могут быть на 30 – 40 % меньше от результатов анализа некипячёной пробы воды, т.е. выпаривание природной воды с целью концентрирования микропримесей может изменять её химический состав, особенно концентрацию лёгких органических веществ.
Метод вымораживания с целью концентрирования в меньшей степени влияет на химический состав пробы воды. Вымораживать воду можно в холодильнике. Следует иметь в виду, что при вымораживании определённое количество растворённых в природной воде веществ переходит в фазу льда, причём чем большая степень концентрирования, тем более значительные потери концентрата. Поэтому вымораживать пробу воды с уменьшением её объема более чем в 8 – 10 раз не рекомендуется.
Ко второй группе принадлежат методы концентрирования, которые базируются на экстракции, сорбции, соосаждении или электрохимическом выделении микрокомпонентов без изменения общего объема пробы воды. Эти методы позволяют не только концентрировать микрокомпоненты, но также отделять их от матрицы и веществ, которые мешают анализу, поэтому они являются более эффективными в сравнении с выпариванием и вымораживанием.
Экстракционное концентрирование состоит в том, что определённый объем природной воды (vвод.) энергично взбалтывают в делительной лейке с меньшим объёмом органического растворителя который практически не смешивается с водой, до установления равновесия. Конечно для этого необходимо не более 3 – 5 минут. Микрокомпоненты в виде органических соединений или комплексов ионов металлов, которые в безводном растворителе растворяются лучше, чем в воде, переходят в органическую фазу. Распределение компонента А между двумя фазами описывается коэффициентом разделения:
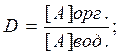 (8)
(8)
Где [А]орг. и [А]вод. – равновесные концентрации А в орг. и водной фазе соответственно.
Если микрокомпонент содержится в водном растворе или в органической фазе в различных молекулярных или ионных формах (например, уксусная кислота в водном растворе содержится в форме НАс и Ас, а в неводном – в виде димеров (НАс)2 и мономеров НАс), то [А]орг. и [А]вод. равны сумме всех форм в каждой из фаз.
Поэтому экстракции с практической точки зрения удобнее характеризовать через степень экстракции R (фактор извлечения), %
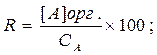 (9)
(9)
Где СА – общая концентрация компонента А в водной фазе до экстракции.
Величины R и D связаны между собой уравнениями
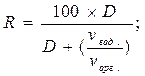
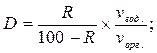 (10)
(10)
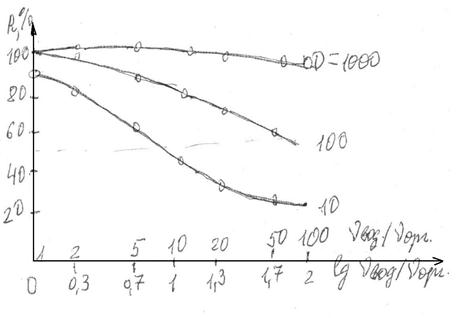
Соотношение объемов водной и органической фаз при практически полной экстракции равняется кратности концентрирования.
Из уравнения (10) вытекает, что степень экстракции зависит не только от значения коэффициента разделения но и от соотношения объемов водной и органической фаз, что показано на рис. 18. Из него видно, что при одноразовой экстракции ≥ 99 % - го извлечения вещества в органическую фазу при кратности концентрирования vвод./vорг. ≤ 10 можно достичь лишь в системах, в которых коэффициент разделения D ≥ 1000. При меньших значениях D или при увеличении отношения vвод./vорг. значительное количество вещества, что распределяется, остаётся в водной фазе.
Во многих случаях доя концентрирования не удаётся найти экстракционных систем с высокими значениями D. В таком случае для практически полного извлечения вещества следует использовать многоразовую экстракцию (например, так делают при экстракционном концентрировании фосфорорганических пестицидов). Расчёт количества экстракций и, необходимых для практически полного извлечения, можно провести по формуле:
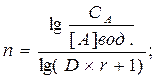
Где СА – общая концентрация вещества А в пробе до экстракции;
[А]вод. – равновесная концентрация в водной фазе после экстракции;
r = vвод./vорг.
Для 99 % - ой экстракции эта формула имеет вид
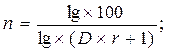
Таблица 10. Зависимость количества экстракций n для 99 % извлечения веществ от коэффициента разделения (D) и кратности концентрирования (vвод./vорг.).
| D | vвод./vорг. | Количество экстракций, n | vвод./vорг. с учётом количества экстракций | |
| Расчёт | Практически | |||
| 0,87 1,00 1,49 1,92 | ||||
| 1,57 1,92 4,19 6,64 | 2,5 5,0 12,5 14,3 | |||
| 4,19 6,64 25,26 48,32 | 1,25 1,43 2,00 2,07 |
Из таблицы видно, что 99 % - го извлечения одноразовой экстракции можно достичь при D ≥ 1000 и vвод./vорг. ≤ 10. Двухразовая экстракция обеспечивает такую же степень извлечения при условии D ≥ 100 и vвод./vорг. ≤ 10.
В других случаях количество экстракций, необходимых для практически полного извлечения веществ, значительно вырастает, что является малоприемлемым с точки зрения техники концентрирования. Кроме того, при увеличении количества экстракций увеличивается и общий объем органической фазы (концентрата). Вследствие этого кратность концентрирования теряется, что приведено в последнем столбце табл. 10
Многоразовую экстракцию можно заменить методом экстракционной хроматографии, т.е. экстракцию в динамических условиях. Для этого органический растворитель наносят на поверхность твёрдого сорбента, которым заполняют сорбционную колонку. При пропускании пробы через такую колонку происходит многоразовое перераспределение веществ, которые экстрагируются, между недвижимой фазой (органическим растворителем) и движимой фазой (водой), что является адекватным многоразовой экстракции. Вследствие этого вещества, что экстрагируются, практически полностью переходят в колонке органическую фазу. Поле этого их можно удалить с колонки малым объемом группового реагента. В случае необходимости использования этой же колонки можно провести хроматографическое разделение сорбированных веществ.
Экстракционное концентрирование применяют в основном для извлечения с природной воды органических соединений и ионов металлов в виде их комплексов с органическими лигандами. Эти инградиенты можно потом определить непосредственно как в экстракте, так и после рестрикции в водной среде.
Сорбционное концентрирование проводят на молекулярных и ионообменных сорбентах в статических и динамических условиях. При концентрировании в статических условиях к пробе природной воды добавляют определённое количество сорбента и выдерживают в течении 20 – 30 минут при интенсивном перемешивании.
Среди молекулярных сорбентов чаще всего используют силикагель, оксид алюминия или активированный уголь. Для сорбции катионов и анионов используют соответственно иониты – катиониты и аниониты.
При концентрировании микрокомпонентов на молекулярных сорбентах изотерма сорбции в большинстве случаев является прямолинейной или близкой к ней и описывается уравнением прямой, которая проходит через начало координат.
QA = Г × СА,
Где Q – количество вещества сорбированного 1 г. Сорбента,
СА – его концентрация в растворе, который находится в равновесии с сорбентом;
Г – коэффициент Генри.
При достаточном излишке сорбента величина Г для данного вещества не зависит от наличия других веществ, в частности от присутствия микрокомпонентов природной воды. После практически полной сорбции микрокомпонентов из большого объёма воды сорбент отделяют фильтрованием и обрабатывают его малым объёмом раствора соответствующего реагента для десорбции. Степень или кратность концентрирования приравнивается отношению объема воды к объему раствора, каким проводилась десорбция.
При концентрировании с помощью ионообменных сорбентов следует учитывать, что состояние равновесия распределения ионов между раствором и сорбентом зависит не только от концентрации ионов, которое поглощается, а также от концентрации протиоионов, которые переходят в раствор при ионном обмене:
Ø концентрирование на катионите в Н+ - форме
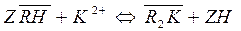 (14)
(14)
Ø концентрирование на анионите в ОН- - форме
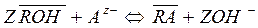 (15)
(15)
Рисками сверху обозначена твёрдая фаза ионообменного сорбента.
Из приведенных уравнений ионообменных реакций видно, что при использовании катионитов и анионитов в Н+ и ОН- - формах коэффициенты распределения ионов зависят соответственно от рН воды, т.е. от концентрации противоионов. На значения коэффициентов раздела влияет также концентрация главных ионов, которые составляют матрицу природной воды и которые могут быть конкурентами в реакциях ионного обмена при недостаточном избытке ионита. Кроме того, состояние равновесий (14), (15) зависит также от массе ионообменного сорбента, который содержит определённую «концентрацию» противоионов. Поэтому коэффициент раздела выражается формулой:
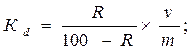
Где R – степень сорбции (извлечения), %;
v – объем природной воды, мл.;
m – масса ионообменного сорбента, г.
При определении коэффициента разделения ионов необходимо указывать концентрацию противоиона, поскольку он влияет на значение Кd.
Коэффициент раздела данного иона не зависит от присутствия других ионов (кроме проитвоионов) при условии, если емкость взятой массы ионообменного сорбента превышает общее количество всех катионов или анионов, которые находятся в природной воде. Емкость большинства синтетических катионов и анионов составляет примерно 5 -10 ммоль экв/г.
В пресных поверхностных водах максимальная сумма катионов или анионов солевого состава составляет ~ 10 – 15 ммоль экв/г. Таким образом, для обеспечения 2 – 3 кратной избыточной емкости ионитов относительно содержания главных ионов с целью избежать их конкуренции при сорбции микрокомпонентов до 1 л. природной воды достаточно добавить 10 – 15 г. ионообменного сорбента. При концентрировании микропримесей ионов с более солоноватых вод количество ионообменного сорбента следует соответственно увеличить
При небольших значениях Кd.концентрирование целесообразно проводить на сорбционных колонках, т.е. в динамических условиях, как при методе экстракционной хроматографии. После сорбции ионов микрокомпонентов из большого объема природной воды их легко десорбировать промыванием катионита концентрированной кислотой или анионита раствором щёлочи. Это обеспечивает практически полное извлечение микрокомпонентов из колонки вследствие сдвига влево равновесий реакции (14), (15). Например, на колонке с катионитом можно сорбировать катионы с 10 л. природной воды, а потом их десорбировать промыванием колонки всего 10 миллиграммами HCl, т.е. достичь степени концентрирования 1000. В случае потребности на этой же колонке можно провести и хромотографическое разделение сорбированных ионов.
При катионобменном концентрировании и разделении следует учитывать, что в природных водах ионы металлов в значительной мере связаны в электронейтральные и анионные комплексы, которые не сорбируются катионитами. Поэтому для обеспечения полной сорбции эти комплексные соединения необходимо предварительно разрушить в использованием различных методов.
Соосаждение применяют в основном для концентрирования микроколичеств ионов металлов. Соосаждение – это процесс захвата микропримесей ионов определённым осадителем в условиях, когда эти ионы сами не образуют малорастворимых соединений. Например, при осаждении сульфата бария с осадителями соосаждается незначительное количество нитрата бария, хотя он является хорошо растворимым соединением; при осаждении гидроксида железа (III) избытком аммиака в присутствии меди к фазе осадка переходит определённое количество ионов Cu2+, хотя они в условиях осаждения образуют хорошо растворимый аммиачный комплекс и т.д. Механизм соосаждения чаще всего заключается в адсорбции или окклюзии микродобавок твёрдой фазой осадка, который называется коллектором. При анализе природных вод применяются различные коллекторы – гидроксид железа, диоксид марганца, гидроксид магния и т.д. Удобным коллектором является гидроксид Mg, который после отделения и растворения практически не мешает дальнейшему определению соосаждённых микрокомпонентов фотометрическим и другими методами. Концентрат, полученный на гидроксиде Mg, можно также анализировать методом эмиссийной спектроскопии, не переводя осадок в раствор.
Инверсная вольтамперометрия с нагромождением является эффективным методом концентрирования и определения микроколичеств металлов в природных водах. Концентрирование ионов металлов осуществляется предварительным электролизом на висящем ртутном точечном электроде (стационарный ртутный электрод) или на твердых электродах (графитовый, ртутно-графитовый, стеклоуглеродный, платиновый, ртутно-серебряный и др.). Электролиз проводят при интенсивном перемешивании раствора в течении 30 – 40 минут при потенциале катода значительно более объемному, чем потенциалы полярографических полуволн определяемых ионов металлов. При этом достигается практически полное выделение и концентрирование на катоде ионов определяемых элементов в виде атомов нулевой степени окисления, твёрдых растворов, химических или интерметаллических соединений и т.д. После окончания концентрирования проводят электрохимическое (анодное) растворение концентрата выделенных металлов с поверхности электрода путём постепенного наложения на электрод сменного потенциала и регистрируют вольтамперную зависимость. На номерограмме «Jдиф – Е» появляются минимумы, потенциалы которых качественно характеризуют природу металла, а глубина минимумов пропорциональна их концентрации. Таким образом, инверсийная вольтамперометрия является одновременно методом концентрирования и качественного определения преимущественно полярографически активных ионов металлов и позволяет проводить многоэлементный анализ.
Удаление веществ, что мешают анализу
Влияния веществ, которые мешают анализу можно избавиться различными методами. Наиболее удобным является способ маскирования, который используется в основном при определении ионов металлов оптическими или электрохимическими методами. Для этого ионы, которые мешают анализу, связывают в стойкие комплексные соединения, которые не дают аналитического сигнала, свойственного определяемому металлу. В то же время добавленный к раствору комплексообразующий реагент не должен влиять на измеряемый аналитический сигнал, т.е. не должен практически связывать определяемый ион. Для выполнения этих условий необходимо чтобы константа стойкости комплексного соединения определяемого иона с маскирующим реагентом была значительно меньшей от константы стойкости иона, который мешает анализу с этим же реагентом.
При анализе природных вод таких условий достичь тяжело через то, что в водах всегда присутствуют неорганические и органические комплексообразующие соединения, влияние которых на ионы, которые определяются и мешают, учесть практически невозможно.
Поэтому для маскирования тех ионов, которые мешают анализу, при возможности их переводят в такую степень окисления, в какой они не мешают анализу, разрушают комплексы или предварительно отделяют ионы металлов экстракционным или электрохимическими методами.
Определению общей концентрации ионов металлов, в частности, d- элементов, особенно мешают органические высокомолекулярные соединения, которые образуют с ионами металлов кинетически инертные стойкие комплексы, которые маскируют элементы, которые необходимо определять (табл. 2 и 3). Поэтому перед определением металлов эти комплексы необходимо разрушить, т.е. учитывать влияние растворённых органических соединений.
В случае, когда ионы металлов определяют в достаточно кислом растворе в виде стойких закрашенных комплексов, например, при определении общего содержания железа в виде комплекса Fe2+ c о- фенатролином (Phen) связывание железа в комплексные соединения FeLn2-h с природными лигандами воды в большинстве случаев не мешает анализу, потому что равновесие
FeLn2-h + 3Phen + nH+ ↔ Fe (Phen)32+ + nHL
Практически полностью сдвигается в сторону образования закрашенной аналитической формы Fe (Phen)32+. В других случаях органические комплексообразующие соединения природных вод предварительно разрушают. Для этого чаще всего используют метод так называемого «мокрого сжигания» или фотохимическую и ультразвуковую деструкцию.
Для мокрого сжигания, которое заключается в окислении органических веществ природных вод, пробу воды подкисляют смесью концентрированной серной и азотной кислот в соотношении 1 : 1 выпаривают до появления густых паров серной кислоты. После этого разбавляют дистиллированной водой, прибавляют раствор KMnO4 и кипятят 3 – 5 минут. Если раствор стал бесцветным, тогда ещё добавляют KMnO4 и опять кипятят. Избыток KMnO4 разлагают кипячением с HCl или добавлением по каплям разведенного раствора Н2С2О4 до обесцвечивания раствора. Для «мокрого сжигания» используют кислоты особой чистоты, в которых содержание тяжёлых металлов незначительно.
При анализе фильтрованной природной воды целесообразно использовать способ фотохимической деструкции органических веществ.
Фотохимическую деструкцию проводят по следующей методике. В пробирку ёмкостью 50 – 100 мл наливают пробы природной воды и добавляют несколько капель разведенной H2SO4 до рН ~ 1. Затем пробы размещают под лучами ртутной лампы мощностью 1000 Вт так, чтобы расстояние от лампы до поверхности воды было не больше 5 – 6 см. Пробирки охлаждают, продувая воздух сбоку вентилятором. Фотохимическую деструкцию проводят в течение 50 – 60 минут. При наличии катализатора HgCl2 деструкция основной массы органических соединений заканчивается за 15 – 20 минут. Полноту деструкции контролируют определением химического употребления О2 (ХУК) отдельной порции воды, обработанной аналогично.
Ультразвуковая обработка проб воды значительно ускоряет разложение органических соединений.
При определении в природных водах органических соединений их при необходимости отделяют от компонентов, что мешают анализу, и концентрируют экстракционным или сорбционным методом.
Автоматизация анализа природных вод
Автоматизация анализа природных вод является одним из важнейших направлений развития методов контроля за состоянием природной среды, особенно в условиях интенсивного промышленного загрязнения природных водных объектов. Актуальность этой проблемы обусловлена тем, что антропогенное загрязнение природных вод во многих случаях имеет бессистемный, периодический, а не беспрерывный характер. Например, в соответствии с технологией очистки промышленные сточные воды часто сосредотачивают в отстойниках, где происходит их частичное очищение вследствие протекания физико-химических и биологических процессов и только после этого воды сбрасываются в речку или водохранилище. Довольно часто в природные водные бассейны неочищенные сточные воды предприятий попадают вследствие аварий на производстве или по причине, что на предприятии нет очистных сооружений или они плохо функционируют. Очевидно, что в таких случаях эффективный контроль за экологическим состоянием водного объекта можно осуществлять только непрерывным или частым периодическим анализом качества природной воды.
Необходимо также иметь в виду, что организация надёжного периодического контроля за химическим составом воды больших водных объектов – водохранилищ, больших озёр и рек – путём отбора десятков и сотен проб во время экспедиций является сложным делом и требует значительных затрат, а осуществление непрерывного и частого периодического контроля за качеством воды в этих объектах вообще невозможно.
Автоматизированный или полуавтоматизированный химический анализ и определение некоторых физических показателей природных вод может выполняться с помощью автоматической станции контроля (АСК) или передвижных гидрохимических лабораторий (ПГХЛ), которые оснащены специальной измерительной аппаратурой.
С помощью помпы и системы отбора проб вода подаётся к блокам первичных преобразователей (детекторов), каждый из которых контролирует один из показателей. Сигналы от первичных преобразователей прямо пропорциональны значениям показателей, которые контролируются. Далее электрические сигналы через многоканальный измерительный преобразователь попадают в блок сбора и обработки информации, который с использованием ЭВМ одновременно выполняет функцию управления всеми блоками автоматического комплекса. Наконец, после обработки полученной информации она может поступать через блок аппаратуры передачи в вычислительный центр, где сосредоточены гидрохимические данные о состоянии всех водных объектов, расположенных в определённом географическом районе.
Блок-схема АСК качества природных вод
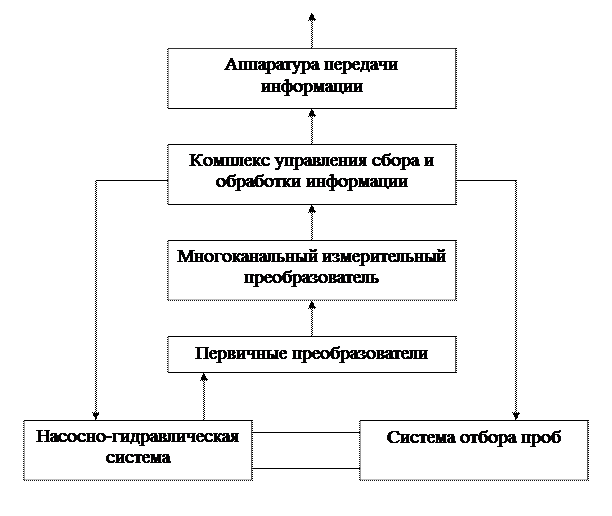
Режим работы АСК может быть стационарным (непрерывным) или периодическим, в зависимости от целей контроля.
Передвижные гидрохимические лаборатории (ПГХЛ) размещаются на специальных грузовых автомобилях с автономным электрохимическим питанием от аккумуляторов или генератора двигателя. В их состав может входить не только электрохимическая и оптическая измерительная аппаратура, которая является основой АСК, а также более сложные анализаторы, например, газожидкостной хроматограф для определения органических соединений, ионный хроматограф для определения катионов и анионов солевого состава воды, атомно-абсорбционный спектрограф для определения тяжёлых металлов и т.д.
Современное развитие науки и техники обусловило возникновение таких перспективных методов контроля за состоянием природных водных объектов как дистанционные и, в частности, аэрокосмические. Они являются чрезвычайно эффективными во время изучения экологического состояния значительных акваторий океанов, морей, больших озёр, водохранилищ и рек.
Методики анализа воздуха
В литературе по анализу воздуха промышленных предприятий приведено несколько сотен методик определения различных химических соединений. Эти соединения можно разделить на следующие группы: металлы и их соединения, неорганические газообразные вещества и органические вещества. Большая часть из них характерна для воздуха производственных помещений и при небольших объемах производства и наличии газоочистных сооружений попадает в атмосферу в незначительных количествах. Поэтому содержание таких веществ в атмосферном воздухе в большинстве случаев не превышает ПДК. В то же время газообразные выбросы многотоннажных производств и энергетических комплексов – такие как оксиды азота, S и C, NH3, HCl, HF и др., пары Hg и особенно токсичные соединения бериллия и свинца, а также некоторые органические соединения – являются экологически опасными даже при локальном загрязнении ими воздушного бассейна.
Металлы и их соединения
Бериллий. Аэрозоли бериллия и его соединений улавливают на фильтре, растворяют и определяют фотометрически по реакции образования закрашенных комплексных соединений с берилоном III или алюминоном. Чувствительность метода составляет 1 и 5 мкг Ве в пробе воздуха.
Статистическая обработка результатов анализа
Основными характеристиками надёжности результатов химического анализа какого-либо объекта являются их правильность и точность (воспроизводимость)
Правильность анализа – это степень адекватности количества (концентрации) определяемого инградиента его действительному содержанию в объекте, который анализируется. Критерием правильности анализа является степень совпадения результатов определения определённого инградиента различными независимыми методами. Например, концентрацию ионов меди в пробе природной воды можно определить методами спектрофотомерии, атомной абсорбции и полярографии. Если средний результат отдельных определений является статистически достоверным, тогда анализ считается правильным.
Такой способ проверки правильности анализа является довольно длительным и громоздким и при анализе природных объектов используется лишь тогда, когда результаты анализа одним методом вызывают сомнения.
Независимо от того, каким способом получено n значений параллельных аналитических измерений, их обрабатывают статистически. Для этого в первую очередь определяют среднее арифметическое значение результатов по формуле:
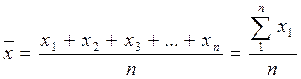 ; (18)
; (18)
Затем вычисляют среднюю квадратичную ошибку (стандартное отклонение) по формуле:
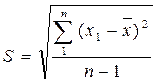 ; (19)
; (19)
и наконец, точность (воспроизводимость) анализа характеризуют величиной доверительного интервала среднего значения, который определяют по формуле:
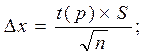
Где t(p) – так называемое t распределение. Его величины для разных значений h при вероятностях Р приведены в таблице.
Зависимость t от n при различных Р
| n | P = 0,90 | P = 0,95 | P = 0,99 |
| 6,31 | 12,7 | 63,7 | |
| 2,92 | 4,3 | 9,92 | |
| 2,35 | 3,18 | 5,84 | |
| 2,13 | 2,78 | 4,60 | |
| 2,01 | 2,57 | 4,03 | |
| 1,94 | 2,45 | 3,71 | |
| 1,89 | 2,36 | 3,50 | |
| 1,83 | 2,26 | 3,25 |
Результаты анализа в любых единицах выражаются следующим образом: 
Значение 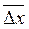 конечно вычисляют с вероятностью Р = 0,95 (95 % - ная вероятность)
конечно вычисляют с вероятностью Р = 0,95 (95 % - ная вероятность)
При определении конкретного инградиента выполняют не более 3 – 4 параллельных анализов, потому что увеличение n незначительно влияет на величину t(p), т.е. на точность анализа.
Из формулы (19) вытекает, что результат единичного измерения является статистически недостоверным, потому что при n = 1 стандартное отклонение S и доверительный интервал среднего значения 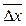 принимают бесконечно большую величину.
принимают бесконечно большую величину.
Важным является определить статистическую достоверность среднего значения 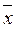 , т.е. убедиться в том, что при выполнении параллельных анализов не было допущено случайной грубой ошибки. При небольших значениях n случайные грубые ошибки находят при помощи разбега варьирования. Для этого рассчитывают соотношение
, т.е. убедиться в том, что при выполнении параллельных анализов не было допущено случайной грубой ошибки. При небольших значениях n случайные грубые ошибки находят при помощи разбега варьирования. Для этого рассчитывают соотношение
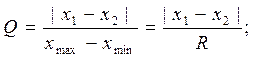 (21)
(21)
Где х1 – значение, которое вызывает сомнение,
х2 – соседнее значение,
R – размах варьирования.
Рассчитанную величину Q сравнивают со значением Q (P,n) в таблице.
| n | P = 0,90 | P = 0,95 | P = 0,99 |
| 0,89 | 0,94 | 0,99 | |
| 0,68 | 0,77 | 0,89 | |
| 0,56 | 0,64 | 0,76 | |
| 0,48 | 0,56 | 0,70 | |
| 0,43 | 0,51 | 0,64 | |
| 0,40 | 0,48 | 0,58 |
Если Q > Q(P, n), то это указывает на наличие грубой ошибки. В таком случае соответствующий результат единичного измерения хi отбрасывают и повторяют расчёт , S и .
Пример. При определении процентного содержания СаО в почве были получены следующие результаты: 2,87; 2,89; 2,90; 2,95. Сомнение вызывает результат 2,95. Проведём расчёты:
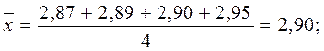
R = 2,95 – 2,87 = 0,08
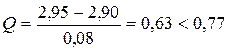
В этом случае Q < Q (P, n), что указывает на отсутствие грубой ошибки, т.е. на статистическую достоверность среднего результата.
Это даёт возможность рассчитать содержание СаО по формулам (18) – (20):

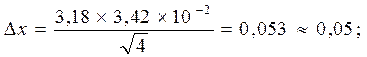
СаО = 2,90 ± 0,05 %
Пример. При определении концентрации ионов аммония в пробе природной воды получены следующие результаты: 7,10; 7,36; 7,40 и 7,42 мг/л
Сомнение вызывает результат 7,10. Проведём расчёты аналогично предыдущим.
= 7,32; R = 7,42 – 7,10 = 0,32;
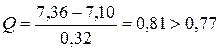
Т.е. результат 7,10 мг/л является грубой ошибкой и его необходимо выключить из дальнейших расчётов. В таком случае
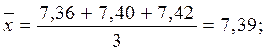
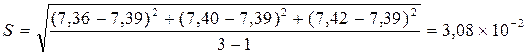
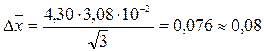 ;
;
CNH+4 = 7,39 ± 0,08 мг/л
Если после исключения грубых ошибок для расчетов остается меньше 3 результатов, то необходимо провести дополнительные анализы.
Особое внимание следует обратить на то, что статистическая обработка результатов анализа, выполненного одним методом, дает возможность обнаружить только случайную ошибку. Если же аналитиком была допущена систематическая ошибка, например, при приготовлении рабочих растворов, при калибровке измерительной аппаратуры и т.д., то обнаружить такую ошибку методом математической статистики невозможно.
Анализ одной пробы, особенно воды или воздуха не может характеризовать с достаточной надежностью ее химический состав, который значительно изменяется во времени и пространстве.
Таким образом, возникает необходимость анализа серии проб, которые отобраны через различные промежутки времени во многих местах исследуемого природного объекта. На основании серийных анализов оценивают средний химический состав исследуемого природного объекта в целом за конкретный промежуток времени. Очевидно, что разбег между результатами серийного анализа многих проб будет всегда значительно большим, чем между результатами параллельных анализов одноразово отобранной пробы. Поэтому при характеристике среднего химического состава воды водохранилищ, речек, озер, воздух большого города, почву с/х угодий и т.д. статистически обрабатывают средние результаты одноразовых анализов. При этом рассчитывают не только средний результат 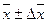 , а также и статически достоверные экстремальные значения содержания конкретного ингардиента хmax и xmin.
, а также и статически достоверные экстремальные значения содержания конкретного ингардиента хmax и xmin.
Для таких расчетов можно применить способ, приемлемый в случае значений 10<n<1000, т.е. для статистической обработки значительного количества результатов анализа. Суть этого способа заключается в том, что отбрасывают крайние значения хі и рассчитывают 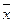 и S. Статистически недостоверными считают результаты, которые отклоняются отна величину, большую чем 4S. После исключения недостоверных результатов рассчитывают , а значения хmax и xmin принимают за достоверные экстремальные величины, характерные для данного природного объекта. Графически эти условия можно изобразить следующим образом:
и S. Статистически недостоверными считают результаты, которые отклоняются отна величину, большую чем 4S. После исключения недостоверных результатов рассчитывают , а значения хmax и xmin принимают за достоверные экстремальные величины, характерные для данного природного объекта. Графически эти условия можно изобразить следующим образом:
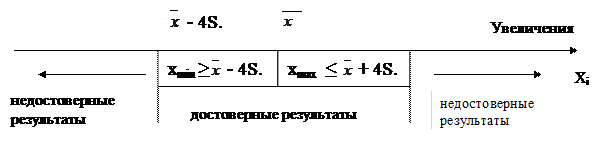
Пример. В воде озера на протяжении одной недели определяли концентрацию ионов Са и по результатам анализа каждой отобранной пробы получили средние значения: 12,0; 10,1; 8,9; 8,3; 8,0; 7,9; 7,8; 7,6 и 6,5 мг/л. Расчеты по формулам (18) – (20) без учета крайних значений дают следующие результаты:
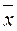 = 8,4; S = 0,81;
= 8,4; S = 0,81;
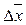 = 0,68
= 0,68 0,7
0,7
Поскольку xmin = 6,5 >- 4S = 5,2 и хmax =12,0> + 4S = 11,6, то результат
xmin является статистически достоверным, а хmax - недостоверным. Статистически достоверным является соседнее значение хmax = 10,1.
Таким образом, средняя концентрация Са2+ и ее экстремальные значения за исследуемый период были такими:
ССа2+ = 8,4 ±0,7 мг/л ССа2+min = 6,5 мг/л ССа2+max = 10,1 мг/л
Подобные результаты часто записывают в виде дроби, указывая в числителе среднюю концентрацию, а в знаменателе – статистически достоверные экстремальные концентрации:
Са2+ = 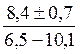 мг/л.
мг/л.
– Конец работы –
Эта тема принадлежит разделу:
Конспект лекций по курсу: Методы и способы измерений параметров окружающей среды ВСТУПЛЕНИЕ
ВСТУПЛЕНИЕ ХАРАКТЕРИСТИКА ОБЪЕКТОВ ПРИРОДНОЙ... ОБЩАЯ СХЕМА АНАЛИЗА И ОСНОВНЫЕ ЭТАПЫ...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: ОТБОР ПРОБ С ВОДНЫХ ОБЪЕКТОВ
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.031 сек.
Новости и инфо для студентов