рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Образование
- /
- Регуляция клеточного цикла. Апоптоз. Онкогенетика.
Реферат Курсовая Конспект
Регуляция клеточного цикла. Апоптоз. Онкогенетика.
Регуляция клеточного цикла. Апоптоз. Онкогенетика. - раздел Образование, ПАВЛИЧЕНКО В. И АБРАМОВ А. В. Митотический Цикл И Его Регуляция. Роль Циклинов И Циклинз...
Митотический цикл и его регуляция. Роль циклинов и циклинзависимых киназ. Принципы передачи митогенного сигнала. Роль факторов роста, интегринов и кадгеринов. Контрольные точки митотического цикла. Апоптоз.
Генетические механизмы канцерогенеза. Общая характеристика генов, берущих участие в канцерогенезе: вирусные онкогены, протоонкогены, гены-супрессоры опухолей, гены-мутаторы. Канцерогенные факторы.
Митотический цикл и его регуляция.Казалось бы, давно известная вещь – фазы цикла (G1, S, G2, M), стадии митоза (профаза, метафаза, анафаза и телофаза), - все уже сто лет как описано и изучено.
Но что заставляет клетку «двигаться» по этому кругу? Как удается ей обеспечить строго упорядоченную смену множества событий, составляющих клеточный цикл? Какие механизмы запускают, например, синтез ДНК в S-фазе, а затем во время его останавливают, предупреждая повторное удвоение ДНК? Как и почему происходит разрушение ядерной оболочки в поздней профазе, а затем образование сразу двух таких оболочек в телофазе? И так далее – подобные вопросы уместны в отношении каждого процесса.
Долгое время все это было совершенно неясно. И только благодаря исследованиям последних лет перед нашим взором стала проступать, с одной стороны, чрезвычайно удивительная (по своей сложности и «продуманности»), а с другой стороны, вполне естественная картина под названием «Регуляция клеточного цикла».
На протяжении клеточного цикла разные клетки могут вступать в различные процессы.
Действительно, делящиеся клетки могут не только продолжать делиться или временно прекратить деления, впав в «спячку» (как бывает со стволовыми клетками). Их судьба, в зависимости от целого ряда обстоятельств (генетической программы, действия гистогормонов, влияние прочих внутренних и внешних факторов), может очень круто измениться. Вот наиболее драматические варианты:
- клетка вступает в процесс дифференцировки,
- в клетке запускаются механизмы самоуничтожения (апоптоза),
- клетка подвергается бласттрансформации, т. е. превращается в опухолевую клетку.
Все это процессы чрезвычайной биологической важности. И то, в какой именно из них вступит делящаяся клетка, определяется в ходе клеточного цикла.
Длительность клеточного цикла изменяется в зависимости от типа клетки и стадии развития организма. Например, клеточный цикл оплодотворенного яйца очень непродолжителен. Оплодотворенное яйцо лягушки делится очень быстро (около 30 минут). Большую часть этого времени занимают S и М фазы, а на фазы G1 и G2 затрачивается очень мало времени. Поскольку в яйце уже существует большой запас белков и других веществ, необходимых для клеточного деления, синтеза новых белков почти не требуется. Таким образом, G1,2- фазы укорачиваются.
На ранних стадиях развития необходимо сформировать как можно больше клеток за относительно короткий промежуток времени. Например, в оплодотворенном яйце лягушки в течение 6 часов происходит 12 дроблений и образуется эмбрион из 8192 клеток. Клетки делятся быстро, но увеличиваются в размерах незначительно. Тем не менее, за счет огромного количества клеток и некоторой ассиметрии деления, эмбрион формируется в течение нескольких часов. После того, как эмбрион сформирован, и закладка трех основных типов ткани завершена, время, необходимое для клеточного цикла, увеличивается за счет удлинения G-фаз. Синтез новых белков приводит к тому, что клетка увеличивается в размере и объеме.
Клеточный цикл оплодотворенного яйца человека, в отличие от яйца лягушки, длится значительно дольше. Первое деление длится 30 часов, а весь процесс дробления – 7 суток (у лягушки – 6 часов).
Однако примерная продолжительность стадий цикла для быстро делящихся клеток человека составляет:
S-период – 10 ч,
G2-период – 4,5 ч,
М-период – 0,5 ч,
G1-период – 9 ч.
Итого – 24 ч (рис.42.). Но, разумеется, это очень приблизительные оценки, и для каких-то клеток цикл может быть более продолжительным. В частности отмечалось, что для сперматогоний S-период длится 15 часов. И, соответственно, в 1,5 раза больше оказывается у них продолжительность цикла – примерно 1,5 суток. Так что на 10 митотических делений сперматогоний (происходящих на первом этапе сперматогенеза) требуется 2 недели.
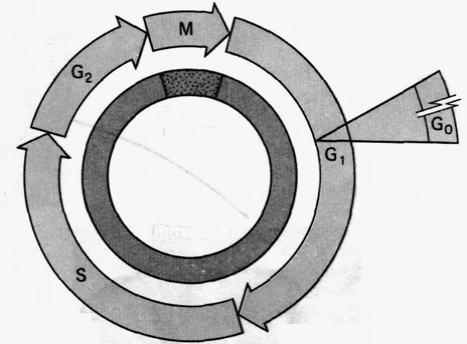
Рис.42. Клеточный цикл: митоз и цитокинез (клеточное деление) составляют М-фазу цикла, кульминацией, которой является образование двух дочерних клеток. Каждая дочерняя клетка вступает в G1-период интерфазы и может начать новый клеточный цикл. За периодом G1 следует S-фаза, во время которой ДНК и хромосомы дуплицируются, и далее – фаза G2. Начало митоза означает конец интерфазы. Покоящиеся клетки задерживаются в фазе G1 и, как говорят, находятся в фазе G0. Обычно эукариотические клетки, которые не остановились в фазе G0, завершают цикл за 24 ч.
Роль циклинов и циклинзависимых киназ.Исследования митотического цикла показали, что ключевую роль в поочередной смене его фаз играют специальные протеинкиназы - т.е. циклинзависимые киназы (Cdks - cyclin-dependent kinasis). Каждая из них фосфорилирует определенные белки, вовлеченные в соответствующую фазу цикла, и таким образом активирует или ингибирует их.
Молекула любой Cdk состоит лишь из одной субъединицы, которая сама по себе неактивна. Для активации же Cdk требуется связывание с ней специального белка - циклина (Ц). Имеется несколько разных циклинов, и, как считают, связавшийся с Cdk циклин не только активирует фермент, но и придает ему субстратную специфичность в отношении тех или иных белков.
Термин циклин отражает тот факт, что концентрации циклинов в клетке в течение клеточного цикла изменяются циклическим образом (рис.43) .
В активной форме протеинкиназы представляют собой гетеродимерные комплексы циклин-Cdk (Ц-Cdk), где циклин служит активаторной, a Cdk - каталитической субъединицей. Данные образования не рассматривают как единые молекулы, а называют комплексами, из-за того, что могут быть разные сочетания конкретных циклинов и конкретных Cdk, причём каждое такое сочетание характерно для строго определённой фазы цикла.
Запускают цикл комплексы циклинD-Cdk4 и (или) циклинD-Cdk6. Разные циклины обозначаются латинскими буквами, а разные Cdk - арабскими цифрами. Названные комплексы функционируют на начальной стадии постмитотического (G1) периода и, вызывая соответствующие внутриклеточные события, способствуют переходу клеткой «точки рестрикции» (точка, предшествующая S-периоду, т.е. для постоянно делящихся клеток находится где-то в конце G1-периода, а для клетки, начинающей деление после перерыва, где-то в конце Go -периода). Аналогично, те же комплексы приводят к возврату «спящей» Go -клетки в митотический цикл.
Вторая половина G1 -происходит под управляющим влиянием комплекса циклин E-Cdk2.
В следующем - синтетическом (S) - периоде функционирующая циклинзависимая киназа остаётся той же (Cdk2), но она дважды меняет своих циклиновых партнёров - ими последовательно становятся циклин А и циклин В. Соответственно, меняются и те белковые субстраты, на которые действуют Cdk2.
В премитотическом (G2) периоде последний из вышеназванных циклинов (В) связывается с другой Cdk - Cdkl. Именно комплекс циклин B-Cdkl «вводит» клетку в митоз и «руководит» этим сложным процессом. Поэтому его еще называют митоз-стимулирующим фактором - MPF (от mitosis-promoting factor).
Из состава последних двух комплексов следует, что их
субстратная специфичность определяется не только циклином, но и самой Cdk, а вернее всего – одновременно тем и другим, т.е. имеющимся сочетанием циклина с Cdk.
Cdk1 нередко обозначается иначе - Cdk2. Дело в том, что мутации гена Cdkl, нарушая клеточный цикл, вызывают у дрожжей фенотип вида Сdc2 (при котором клетки становятся необычайно длинными). Поэтому, ещё до идентификации функции этого гена, он был обозначен как Сdc2.
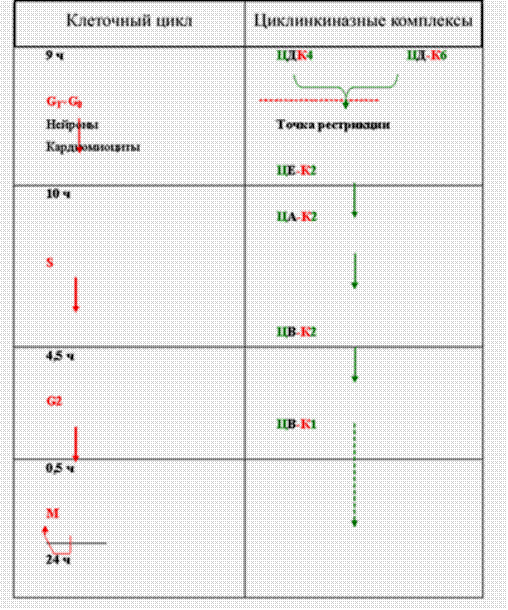
Рис.43. Схема динамики клеточного цикла
Итак, всё разнообразие событий клеточного цикла управляется относительно небольшим числом комплексов циклинов и Cdk.
Способы регуляции и активности Cdks. Ввиду исключительной функции циклинзависимых киназ (Cdks), их содержание и особенно активность находится под сложным контролем. Вот принципиальные способы этого контроля.
1. Регуляция синтеза Cdks. Видимо, не все Cdks одновременно присутствуют в клетке на разных стадиях её цикла. Поэтому очень важным моментом является активация гена той или иной Cdk.
Так, известно, что комплексы G1 периода (циклин D-Cdk4,6 и циклин E-Cdk2), помимо других многочисленных действий, запускают транскрипцию гена киназы Cdkl. Последняя же необходима для образования комплекса G2- периода и митоза (циклин B-Cdkl или MPF). Таким образом, о синтезе компонентов данного комплекса клетка «заботится» заблаговременно, ещё до репликации ДНК.
2. Регуляция активности Cdks. Способы этой регуляции достаточно разнообразны.
а) Один из них - связывание с какой-либо Cdk активаторной
субъединицы - циклина.
б) Второй способ - связывание (либо только с Cdk , либо со всем комплексом циклин-Cdk) ингибиторной субъединицы. В роли последней выступают специальные белки (р15, р16, р21, р27, р57). Известно, по крайней мере, два семейства таких белков. Белки семейства INK4 (р15 и р16) связываются с Cdk4,6 и тем самым препятствуют образованию комплексов (циклин D-Cdk4,6), запускающих клеточный цикл. Белки второго семейства, KIP1 (р21,р27, р57), связываются с уже сформированными комплексами - и это тоже приводит к ингибирующему эффекту.
с) Третий способ контроля активности Cdks - это их фосфорилирование и дефосфорилирование. Т.е. данные протеинкиназы (Cdks) сами способны регулироваться тем же самым способом, каким они управляют активностью «подведомственных» им белков.
Фосфорилирование по одним локусам Cdk обладает активирующим действием; фосфорилирование других участков, напротив, ингибирует фермент.
Но активность киназ восстанавливается под действием специфической фосфатазы, кодируемой геном cdc25a. Из обозначения следует, что это ещё один ген, мутация которого у дрожжей приводит к нарушению клеточного цикла и фенотипу cdc.
Выявлены также две тирозинкиназы (ТК), ингибирующие другую Cdk - Cdkl; одна из этих ТК, видимо, действует на свободную Cdkl, a другая - на весь комплекс циклин В -Cdkl.
3.Регуляция синтеза и распада активаторов и ингибиторов Cdks. В качестве примера обратимся вначале к циклинам.
а) Многие митогенные факторы, «побуждая» клетку к делению, запускают такие регуляторные цепи, которые, в конце концов, активируют ген циклина D (помимо многих других действий). На следующих стадиях клеточного цикла стимулируется синтез очередных циклинов и т.д.
б) Крайне интересным является также управление распадом циклинов. Оно осуществляется с помощью убиквитин зависимого механизма, который используется для короткоживущих белков.
Наиболее чётко роль этого механизма показана для циклина В, участвующего в образовании комплекса циклин В - Cdk2 и митоз-стимулирующего фактора (MPF, он же комплекс циклин B-Cdkl). Фосфорилируя определённые белки, MPF стимулирует вхождение клетки в митоз. Но для завершения митоза необходимы во многом прямо противоположные события. Т.е. содержание MPF должно снижаться. Это достигается с помощью быстрого распада циклина В.
Примерная последовательность событий. Максимальной активности комплекс MPF достигает в метафазе митоза. В это время он фосфорилирует, помимо других белков, так называемый фактор АРС (аnaphase-promotoring complex) т.е. фактор, обеспечивающий анафазу. А данный фактор является ничем иным, как убиквитинлигазой, специфичной в отношении MPF. Поэтому он начинает последовательно присоединять (одну за другой) молекулы убиквитина к циклину В. Меченный таким образом циклин В быстро разрушается в протеосомах. В итоге содержание комплекса циклин B-Cdkl значительно снижается, и в клетке благополучно совершаются события анафазы, а затем и телофазы. Затем в G1-периоде фактор АРС инактивируется, от чего скорость распада циклина В снижается, и данный циклин начинает накапливаться в клетке.
в) Контроль за синтезом веществ, ингибирующих Cdks (либо комплексы циклин-Cdk). Регуляция синтеза часто используется внеклеточными эффекторами для влияния на клеточный цикл - стимуляции или торможения пролиферации. Один из примеров: в некоторых регуляторных внутриклеточных цепочках фигурируют белки семейства Smad. Эти белки, образуя соответствующие транскрипционные факторы, стимулируют синтез ингибиторов р15, р21 и др. В итоге активность комплексов G1-периода тормозится и клетка прекращает деление.
Принципы передачи митогенного сигнала. Практически все сигнальные пути, регулирующие пролиферацию клеток, «нацелены» на комплексы G1-периода — в основном, циклин D—Cdk4,6 и, в меньшей степени, циклин Е—Cdk2. Это и понятно: как уже отмечалось, именно данные комплексы «запускают» очередной клеточный цикл.
Рассмотрим два митогенных сигнальных пути. Один из них относится к стволовым клеткам эпителия и начинается со связывания ЭФР — эпидермального фактора роста.
Второй путь активируется в Т-хелперах после их взаимодействия с АПК — антигенпредставляющими клетками.
Теперь на этих же двух примерах покажем «выход» подобных путей на комплексы циклин-Cdk.
В обоих случаях внешний сигнал приводит к активации
тирозинкиназы, ассоциированной с рецептором. Это ведет (через те или иные посредники) к запуску каскадов митогенактивируемых протеинкиназ (МАПК). Конечные ферменты данного каскада, фосфорилируя ряд транскрипционных факторов (Elk, Ets, ATF2, Tcf и др.), активируют их, а через них — и т. н. гены раннего ответа (FOS, JUN). В культуре клеток уже через 30 мин после начала действия митогена активность этих генов достигает максимального
уровня.
Продукты семейств генов FOS и JUN — это опять-таки
транскрипционные факторы, но специфичные уже в отношении других генов — т. н. генов замедленного ответа. Поэтому через некоторое время начинается экспрессия этих генов.
Среди последних-то и находятся наши «знакомые» — гены циклина D, Cdk4 и Cdk6, т.е. компонентов комплексов циклин-Cdk, специфичных для первой половины G1 - периода цикла.
Кроме того, активируются еще некоторые гены и среди них — ген белка Мус.


 После своего синтеза белок Мус, в свою очередь, влияет на активность ряда генов. Так, он тормозит экспрессию гена белка р27 — ингибитора целого ряда комплексов циклин- Cdk. И одновременно белок Мус активирует ген Cdc25a, который кодирует специфическую фосфатазу. Последняя дефосфорилирует Cdk4 и Cdk2, что приводит к их активации.
После своего синтеза белок Мус, в свою очередь, влияет на активность ряда генов. Так, он тормозит экспрессию гена белка р27 — ингибитора целого ряда комплексов циклин- Cdk. И одновременно белок Мус активирует ген Cdc25a, который кодирует специфическую фосфатазу. Последняя дефосфорилирует Cdk4 и Cdk2, что приводит к их активации.
В результате мы получаем следующие эффекты:
увеличение содержания в клетке циклина D и реагирующих с ним киназ — Cdk4, Cdk6;
снижение содержания ряда ингибиторов Cdks;
повышение активности (в результате дефосфорилирования) тех же циклинзависимых киназ (Cdk4, Cdk6), а также Cdk2.
Все это и обеспечивает формирование в клетке достаточного количества активных комплексов циклин D—Cdk4,6, начинающих подготавливать клетку к делению.
Заметим, чтобы рассмотренные сигнальные пути «сработали», необходимо одно условие: клетка должна быть фиксирована на каком-либо внеклеточном матриксе.
Действие антимитогенов. Это действие ФНО (фактора некроза опухолей) на опухолевые клетки. ФНО «запускает» в клетках сразу несколько сигнальных путей, и среди них один содержит ферментативный каскад МАПК. Только теперь возбуждение соответствующего рецептора через ряд посредников (среди которых — сфингозин и ПК-С) приводит к торможению МАПК. Дальше — все как в предыдущих путях, но с противоположным знаком. Следовательно, в итоге в клетке будет резко снижаться количество активных комплексов циклин D—Cdk4,6, в результате чего деления прекратятся. Действие же ФНО через другие сигнальные пути будет инициировать апоптоз.
Второй пример: речь идет о ТФРр — трансформирующем факторе роста. Уже отмечалось, что он угнетает пролиферацию многих клеток. Рецепторы ТФРр, видимо, сходны по структуре с рецепторами ЭФР (эпидермального фактора роста). Это значит, что при связывании своего лиганда рецепторные субъединицы объединяются в димерные структуры, цитоплазматические домены каждой субъединицы обладают тирозинкиназной активностью, в активированном состоянии эти домены фосфорилируют друг друга. Отличие состоит лишь в том, что в данном случае модифицированные домены рецептора связывают иной комплекс цитоплазматических белков: теперь это комплекс Smad2+Smad3. Он тоже фосфорилируется рецепторной тирозинкиназой, после чего приобретает способность связывать третий белок — Smad4.
Затем весь этот тройной комплекс диффундирует в ядро и выполняет роль транскрипционного фактора для генов, кодирующих ингибиторы Cdks. Имеются в виду такие белки, как р15 и р21.
Накопление в клетке этих ингибиторов и приводит к торможению пролиферации.
Роль факторов роста, интегринов и кадгеринов. Клеткам низших организмов для вступления в S-фазу клеточного цикла необходимо наличие питательных веществ во внешней среде. При недостатке питательных веществ клетка не делится. У высших организмов наличие питательных веществ обычно не является лимитирующим фактором. Однако сигнал, инициирующий деление клетки, чаще бывает внешним, чем внутренним. Доказательства этого были получены в первых экспериментах с культурой клеток млекопитающих in vitro.
При разработке методики выращивания клеток млекопитающих в культуре было отмечено, что клетки растут лучше, если они находятся внутри кровяных сгустков. Из-за неудобства исследования растущих клеток внутри сгустка, после свертывания крови сгусток удаляется. Оставшаяся жидкость известна как сыворотка. Если удалить все клетки до того, как кровь свернется, и добавить полученную сыворотку к растущим клеткам, она не будет поддерживать рост. Это доказывает, что при свертывании из клеток крови высвобождается некий фактор, необходимый для деления клеток. Было показано, что это вещество высвобождается из тромбоцитов, и оно было названо тромбоцитарным фактором роста (PDGF). Оказывается, нормальные клетки млекопитающих содержат достаточное количество питательных веществ для деления. Таким образом, в клетки должен поступать дополнительный сигнал, стимулирующий пролиферацию. В случае с тромбоцитарным фактором роста этот белок секретируется тромбоцитами при свертывании крови после образовании раны. Для восполнения тканевого дефекта тромбоцитарный фактор роста, один из главных факторов роста организма, сигнализирует окружающим клеткам о необходимости приступить к делению.
К настоящему времени выделено и охарактеризовано большое число факторов роста: тромбоцитариый фактор роста; эпидермальный фактор роста (EGF); факторы роста фибробластов (FGF) (имеют девять изоформ и обладают небольшой клеточной специфичностью, однако еще относятся к группе малоспецифичных); фактор роста нервов (NGF) (этот фактор роста действует только на клетки нервной системы); эритропоэтин (ЕРО) (этот фактор роста стимулирует образование эритроцитов в костном мозге); иитерлейкин-2 и интерлейкин-3.
Все эти факторы роста действуют в основном на специфические клетки-мишени. Каждый фактор роста является лигандом, который связывается со специфическим поверхностным рецептором клетки и инициирует процесс передачи сигнала.
Большинство клеток млекопитающих находится в особой фазе клеточного цикла, называемой Go-фазой или компартментом клеточной дифференцировки. Эта фаза клеточного цикла представляет собой удлиненную фазу G1. Это означает, что гены, кодирующие белки, которые запускают пролиферацию клеток, находятся в «выключенном» состоянии. В этом положении метаболическая энергия клетки расходуется на образование специализированных белков, необходимых для осуществления дифференцировки. Таким образом, гены, кодирующие эти белки, находятся во «включенном» состоянии.
В большинстве типов клеток гены для клеточной пролиферации могут быть включены снова. Однако в некоторых клетках гены пролиферации никогда не запускаются повторно после выключения. При завершении пролиферации в клетках сердечной мышцы Cdk и циклины выключаются и больше никогда не включаются. То же справедливо и для всех типов нейронов.
Однако в соответствующих условиях многие клетки могут вернуться в цикл деления. Практически во всех тканях организма происходят клеточные «превращения». Обычно такие превращения происходят редко. Исключение составляют клетки крови и кишечного эпителия, в которых скорость обновления клеток очень высока.
Как показано выше, один из основных типов сигналов, активирующих Cdk и циклины, генерируют факторы роста. Однако, даже получив такой сигнал, организм должен регулировать ответ таким образом, чтобы поддерживать количество и пространственную организацию клеток. Если этого не происходит, наступают летальные изменения организма.
При исследовании нормальных клеток, выращенных в культуре ткани, было показано, что существуют различные факторы, определяющие число клеток, необходимых для поддержания архитектуры ткани, и качество соединения клеток с базальной мембраной. Если выращиваемые в культуре фибробласты заполняют всю чашку Петри, они перестают делиться и, очевидно, уходят в псевдо-Со-фазу. Если в стерильных условиях по поверхности культуры провести шпателем диагональную линию, клетки, расположенные на этой линии, погибают и остается бесклеточная диагональ. Клетки, находящиеся на краю «раны», начинают немедленно делиться и пролиферировать, заполняя свободное пространство. Как только пространство заполнено, деление снова прекращается.
На основе этого наблюдения был сделан вывод о том, что нормальные клетки делятся до достижения определенной плотности. После образования контактов с другими клетками на субстрате, внутрь клетки передается сигнал о том, что все контактные точки заняты, и гены, запускающие пролиферацию, выключаются.
Если клетке не давать соприкоснуться с поверхностью тарелки, она сохраняет округлую форму и не вступает в митоз. Если же клетка соприкасается с поверхностью и расплющивается, она формирует с поверхностью тарелки контакты, называемые фокальными контактами и фокальными адгезионными пластинками. В состав фокальной адгезионной пластинки входят важные компоненты цитоскелета, включая актиновые филаменты, микротрубочки и другие структурные белки. Спустя некоторое время эти клетки вступают в митоз.
Результаты этих и аналогичных экспериментов доказывают, что молекулы окружающей среды, например входящие в состав межклеточных контактов и контактов клеток с межклеточным веществом, участвуют в управлении клеточной пролиферацией.
Теперь рассмотрим такие сигнальные пути, которые начинаются от адгезивных мембранных белков. Одни из этих путей стимулируют пролиферацию клеток; другие, наоборот, тормозят.
Многие клетки способны делиться, только будучи прикрепленными к внеклеточной структуре – базальной мембране (эпителиоциты), коллагеновым волокнам (фибробласты) и т.д.
Информация же о связи клетки с такой структурой поступает от интегринов. Это адгезивные белки с двумя неравными субъединицами.
При связывании с внеклеточным матриксом меньшая (р-) субъединица интегрина активирует одну из тирозинкиназ — FAK (Focal Adhesion Kinase), а та, в свою очередь, еще одну тирозинкиназу — Src. Последняя относится к классу нерецепторных тирозинкиназ, поскольку не входит в состав рецепторного комплекса и даже не взаимодействует напрямую с рецепторами.
Непосредственным субстратом Src является белок SHC, который связывает данный сигнальный путь (идущий от интегринов) с сигнальными путями от рецепторов митогенов. Общей же частью последних путей являются многократно упоминавшиеся выше каскады МАПК (митогенактивируемых протеинкиназ). Следовательно, адаптерный белок SHC должен стимулировать прохождение сигнала по данным каскадам.
Однако не вполне ясно, какой именно член каскадов активируется белком SHC. Есть предположение, что это тот же комплекс белков (GRB, SOS, Ras, Raf), что и в случае действия ЭФР (эпидермального фактора роста).
Но, с нашей точки зрения, надо учесть следующее обстоятельство. Для того, чтобы клетка млекопитающих стала делиться, необходимо одновременное выполнение двух условий: прикрепление клетки к какой-либо поверхности и действие на нее ростового фактора.
Причем в неприкрепленной клетке прохождение сигнала от ростового фактора блокируется на одной из МАПК, а конкретно — на киназе МЕК (МЕК1).
Отсюда следует, что:
а) сигнал от интегринов идет именно к этому звену МАПК — киназе МЕК;
б) данная киназа активируется только при действии на нее сразу двух белков: предыдущей киназы из семейства МАПК и белка SHC (после действия на него Src).
Тогда и получается, что сигналы от ростового фактора и интегринов взаимно дополняют друг друга, а каждый из них в отдельности не способен индуцировать деление клетки.
Эффект прикрепления клетки к опоре включает еще одно важное событие. Оно касается «знаменитого» транскрипционного фактора — белка р53.
Последний активирует гены, останавливающие деления (в частности, ген белка р21 — ингибитора комплексов циклин- Cdk), а также гены апоптоза. Поэтому неудивительно, что идущий от интегринов сигнал приводит к снижению содержания в клетке такого «вредного» белка.
В случае белка р53 обычно регулируется не синтез, а распад. Поэтому под действием сигнала от интегринов, видимо, тем или иным способом ускоряется протеолиз этого белка. По некоторым данным, это может быть связано со снижением содержания другого белка (ARF), ингибирующего распад белка р53.
Контактное торможение пролиферации. Если же клетка устанавливает контакт не с внеклеточным матриксом, а с другими клетками, то наблюдается эффект, прямо противоположный предыдущему, — прекращение делений. Это обозначается как контактное торможение.
После очередного пересева клеток в плоский стеклянный сосуд («матрас») они вначале — в течение нескольких часов прикрепляются ко дну сосуда и лишь после этого входят в клеточный цикл (стимуляция делений).
Когда же на дне сосуда не остается свободного места и клетки вступают в контакт друг с другом, деления прекращаются (контактное торможение).
Сигнал о межклеточном контакте, видимо, идет от кадгеринов — адгезивных белков, участвующих в образовании таких контактов. Вероятно, у кадгеринов меняется при этом конфигурация; поэтому их цитоплазматические домены приобретают способность связывать белок р-катенин.
Предполагают, что данный белок — транскрипционный фактор. Когда он свободен от связи с кадгерином (т. е. когда клетка еще не контактирует с другими клетками), он образует активный комплекс с еще одним транскрипционным фактором — белком Tcf4. Комплекс мигрирует в ядро и здесь прямым или (скорее всего) опосредованным способом стимулирует транскрипцию генов циклина D и белка Мус.
Это вновь те самые гены, которые активируются в ранее рассмотренных сигнальных путях — идущих от ростовых факторов и от интегринов. Те пути сходятся и взаимно дополняют друг друга на каскадах митогенактивируемых киназ (МАПК).
Но путь, содержащий β-катенин, тоже является необходимым для деления. При улавливании β-катенина кадгерином деления прекращаются. Следовательно, этот путь также необходимо дополняет предыдущие. С нашей точки зрения, все объясняется и встает на свои места при следующих предположениях:
· в начале любого клеточного цикла должен образоваться некий (всегда один и тот же) промитотический комплекс транскрипционных факторов (ПМКТФ), специфичный в отношении генов раннего ответа;
· в состав этого комплекса входят β-катенин и белок Tcf4, что возможно лишь тогда, когда β-катенин не связан кадгерином (нет межклеточных контактов);
· кроме того, комплекс содержит факторы, активируемые каскадом МАПК; причем этот каскад сам, по предыдущему нашему предположению, требует двойного сигнала от интегринов (клетка прикреплена к внеклеточной структуре) и от ростового фактора.
В итоге число условий, необходимых для деления клетки (в принципе способной к делению), возрастает уже до трех: это не только прикрепление к поверхности и действие ростового фактора, но и отсутствие контактов с другими клетками.
Лишь при выполнении этих условий активируются гены раннего ответа, и запускается механизм клеточного цикла, в частности, образуется «вторая очередь» транскрипционных факторов, а затем и продукты стимулируемых ими генов — циклин D, Cdk4,6 и т. д.
Но этим перипетии внутриклеточных регуляторных путей, связанные с β-катенином, не исчерпываются. Установлено еще, по крайней мере, два интересных факта.
а) Деградация β-катенина производится с помощью убиквитинзависимого механизма. Причем в качестве убиквитинлигазы (фермента, переносящего убиквитин на Р-катенин) выступает уже знакомый нам фактор АРС, обеспечивающий анафазу.
Последний выполняет такую же функцию в отношении циклинов, необходимых для вхождения клетки в цикл. Поэтому нет ничего удивительного, что одновременно с данными циклинами разрушается и транскрипционный фактор, требующийся для их синтеза. Происходит же это в метафазе и анафазе митоза, когда АРС активируется путем фосфорилирования.
По некоторым данным, перед взаимодействием с АРС β-катенин тоже должен подвергнуться фосфорилированию — киназой-ЗР-гликогенсинтетазы (GSK-3P), которая, в свою очередь, находится под контролем ряда белков.
Такое «повышенное внимание» со стороны клетки к β-катенину — косвенное свидетельство его ключевой роли в запуске деления. Это вполне согласуется с предположением, что β-катенин — необходимый компонент универсального транскрипционного комплекса, начинающего клеточный цикл при любом митогенном стимуле.
б) Второй факт состоит в том, что при контактном торможении в клетке увеличивается содержание белка р53. Это тоже выглядит вполне естественно, если считать, что все тот же комплекс транскрипционных факторов, помимо прочего, ускоряет распад белка р53, выключая ген белка ARF — ингибитора протеолиза белка р53.
Конечно, известно здесь еще далеко не все. Но о принципе работы и некоторых деталях этого замечательного механизма уже можно сказать.
Что касается принципа работы, то он достаточно прост и очевиден. Комплекс циклин-Cdk очередной стадии цикла, как правило, должен обеспечить три процесса:
а) «выведение из игры» комплекса предыдущей стадии,
б) стимуляцию событий «своей» стадии,
в) образование (или активацию) комплекса следующей стадии.
Получается своего рода «цепной» механизм, после включения, которого каждая стадия процесса подготавливает условия для перехода к следующей стадии. Несомненно, таков же принцип многих других сложных биологических процессов — таких, как эмбриональное развитие, дифференцировка, регенерация.
Посмотрим теперь, как конкретно реализуется этот принцип в случае клеточного цикла.
Контрольные точки митотического цикла. (Белок р53, микротрубочки: связь с клиникой). Точная репликация и распределение генетического материала — это важнейшие условия выживания клетки. В клеточном цикле существуют четыре точки, в которых точность репликации, правильность последовательности и равное разделение ДНК контролируются специальными клеточными механизмами.
A. Контрольная точка фазы G1. Если в фазе G1 обнаруживается повреждение ДНК, белок р53 выступает в роли фактора транскрипции и вызывает задержку клеток в G1. Нестабильный р53 обычно быстро разрушается. Однако, когда в клетке появляется аномальная ДНК, белок р53 стабилизируется и присоединяется к этой ДНК. В результате р53 накапливается в ядре и стимулирует экспрессию белка, ингибирующего Cdk2.
Клетка задерживается в фазе G1 до тех пор, пока поврежденные нуклеотиды не будут восстановлены ферментами репарации. Задержка в фазе G1 предотвращает копирование поврежденных оснований и тормозит мутацию ДНК. При отщеплении белка р53 от ДНК его концентрация снижается; ингибитор Cdk отделяется, и Cdk начинает экспрессироваться.
В клетках многих метастатических опухолей человека оба аллеля р53 неактивны. В таких клетках нарушен нормальный контроль, осуществляемый р53, и поврежденная ДНК может реплицироваться. В некоторых случаях это приводит к метастатической трансформации опухолей.
Б. Контрольная точка S-фазы функционирует в фазе S, когда реплицируется ДНК. Появление мутаций в процессе репликации и их последующее встраивание в геном может вызвать серьезные последствия, включая гибель клетки. Если произошли ошибки в репликации, (что случается) и если они были пропущены репаративными ферментами, клетка не может выйти из S-фазы. Проверка точной репликации ДНК — важнейшая регуляторная точка клетки.
B. Контрольная точка С2-фазы. Нереплицированная ДНК блокирует переход клетки от С2 -фазы к М-фазе. Происходит катастрофическое повреждение, если клетка проходит через фазу цикла, незавершив молекулярные процессы, необходимые для подготовки к делению. Например, при введении MPF в клетки, находящиеся в S-фазе, неполностью реплицированные хромосомы конденсируются, а затем фрагментируются.
Г. Контрольная точка М-фазы. Причиной остановки цикла в данной точке может быть неправильная сборка веретена деления. Например, неприкрепление кинетохоры какой- либо хроматиды к микротрубочкам веретена деления. Микротрубочки (МКТ) постоянно полимеризуются и деполимеризуются. Во время митоза происходят резкие и непредсказуемые переходы МКТ из растущего (полимеризация) в сокращающееся (деполимеризация) состояние - это катастрофа и обратно – это спасение. Медикаментозная терапия онкологических больных заключается в разрушении митотического веретена колхицином, винбластином и винкристином, которые нарушают полимеризацию МКТ.
В зависимости от результатов «проверки», выбирается один из трех вариантов:
а) безостановочный переход к следующей стадии цикла;
б) задержка на текущей стадии для исправления обнаруженных дефектов;
в) запуск механизма апоптоза, если выявленные нарушения неисправимы.
В большинстве, если не во всех, случаях хромосомных повреждений центральную роль в остановке цикла играет белок р53.
Роль белка р53 («Страж генома», «Диспетчер апоптоза», Опухолевый супрессор). Белок р53 контролирует исключительно важные клеточные процессы и, благодаря этому, вовлечен в большое количество всевозможных регуляторных цепей. Он (или его ген) активируется в ответ на разнообразные повреждения клеточной структуры: нерепарированные разрывы и другие повреждения ДНК, нарушение расхождения хромосом в митозе, разрушение микротрубочек и т. д.
Сам же белок р53 регулирует активность, по крайней мере, трех групп генов:
1) активирует гены (Р21, GADD45 и другие), отвечающие за остановку клеточного деления;
2) активирует гены (BAX, KILLER/DR5, PIG и другие), запускающие апоптоз – процесс, ведущий, путем активации специальных ферментов, к гибели клетки; а так же репрессирует гены (BCL2, RELA), сдерживающие апоптоз;
3) активирует гены (TSP1, BAI1 и другие), тормозящие ангиогенез (образование новых сосудов).
В итоге через посредничество белка р53 клетка в ответ на повреждение своей структуры
- либо задерживается на той или иной стадии митотического цикла и исправляет эти повреждения;
- либо (при невозможности исправлений) вообще прекращает деления и вступает в процесс клеточного старения (фаза III по Хейфлику);
- либо (при потенциальной опасности поврежденной клетки для ее окружения) осуществляет апоптоз, т. е., попросту говоря, самоубийство.
В частности, апоптозу, помимо прочих, подвергаются и клетки, в которых произошла опухолевая трансформация. В этой связи понятно, почему одновременно тормозится ангиогенез: это еще один способ ограничения опухолевого роста.
Поэтому белок р53 – один из наиболее важных опухолевых супрессоров. В большинстве уже развивающихся опухолей функции белка р53 оказываются в том или ином отношении нарушены.
Такова общая биологическая роль белка р53, а теперь рассмотрим некоторые связанные с ним вопросы более детально.
Белок р53 состоит из 392 аминокислот, образующих 6 доменов (рис. 44):
1) N-концевой домен активирует ген белка р21 (ингибитор киназ), а так же других белков, останавливающих деление клетки.
2) дополнительный транскрипционный домен для активации генов-мишеней.
3) гибкий пролиновый домен проявляет супрессорную активность, запускает апоптоз.
4) центральный домен связывается с энхансерами. В этом участке мутации 5-8 экзонов приводят к онкогенезу.
5) α-спиральный домен отвечает за ядерную локализацию р53 и образование тетрамеров (мономеры не активны).
6) С-концевой домен – мишень для модифицирующих ферментов (киназ, ацетилаз, гликозилаз).


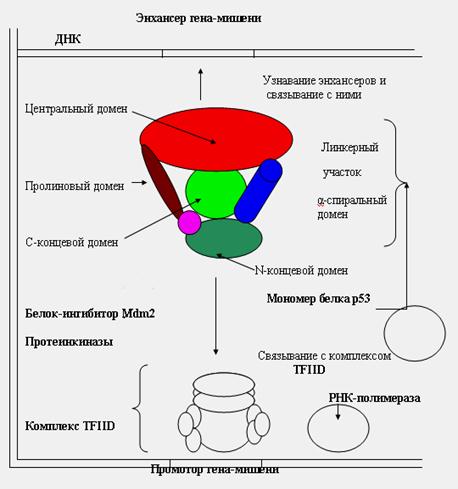
Рис.44. Белок р53
Если С-концевой домен не модифицирован, центральный домен не способен взаимодействовать с ДНК-мишенью. Модификация же С-домена не только придает белку р53 такую способность, но и влияет на его специфичность. Дело в том, что р53-зависимые энхансеры (относящиеся к разным генам) несколько различаются последовательностью нуклеотидных пар. И от вида модификации С-домена зависит, с какими конкретно энхансерами будет связываться белок р53, а с какими – нет.
Таким образом, с помощью модификации двух концевых ( N- и С-) доменов белок р53 получает (от очень многочисленных «источников») информацию о состоянии клетки, перерабатывает ее путем изменения своей конфигурации и надлежащим образом реагирует как транскрипционный фактор определенных генов.
С-концевой домен выполняет еще одну функцию. Как отмечалось выше, некоторые гены (BCL2, RELA) белком р53 не активируются, а репрессируются. Это действие, как считают, осуществляется С-доменом. При этом последний (вместо N-домена) связывается с комплексом TFIID и подавляет его активность.
Апоптоз (Программированная гибель клеток).В Англии в Кембриджском университете в начале 60х годов Сидней Бреннер изучал нематоду Сaenorzhabditis elegans – 1мм длиной, прозрачна (видно деление клеток). Сотрудник его лаборатории Джон Салстон описал схему развития из яйцеклетки взрослого червя. При развитии нематоды образуется 1090 клеток, из которых 131 клетка идет в апоптоз, остается 959 клеток. Гены смерти, приводящие клетки к апоптозу, установил американец Роберт Хорвиц. Впоследствии, благодаря этим работам, они стали лауреатами Нобелевской премии в области молекулярной биологии.
Термин «апоптоз» появился в науке в 1972г. I. Kerr заимствовал его у Гиппократа, в переводе с греческого «апоптоз» – осенний листопад.
Апоптоз - программированная клеточная гибель, энергетически зависимый, генетически контролируемый процесс, который запускается специфическими сигналами и избавляет организм от ослабленных, ненужных или повреждённых клеток. В организме здорового человека клеточный гомеостаз определяется балансом между гибелью и пролиферацией клеток. Ежедневно, примерно около 5% клеток организма подвергаются апоптозу, а их место занимают новые клетки. В процессе апоптоза клетка исчезает бесследно в течение 15-120 минут. Апоптоз – это биохимически специфический тип гибели клетки, который характеризуется активацией нелизосомных эндогенных эндонуклеаз, которые расщепляют ядерную ДНК на маленькие фрагменты. Морфологически апоптоз проявляется гибелью единичных, беспорядочно расположенных клеток, что сопровождается формированием округлых, окруженных мембраной телец (“апоптотические тельца”), которые тут же фагоцитируются окружающими клетками.
Это энергозависимый процесс, посредством которого удаляются нежелательные и дефектные клетки организма. Он играет большую роль в морфогенезе и является механизмом постоянного контроля размеров органов. При снижении апоптоза происходит накопление клеток, пример – опухолевый рост. При увеличении апоптоза наблюдается прогрессивное уменьшение количества клеток в ткани, пример – атрофия.
Назначение апоптоза в клеточных популяциях можно сформулировать таким образом:
· поддержание численности клеток в популяции на заданном уровне;
· определение этого уровня и его изменение под влиянием внешних (по отношению к клетке) сигналов вплоть до полной элиминации данного типа клеток;
· селекция разновидностей клеток внутри популяции (в том числе элиминация клеток с генетическими дефектами).
Морфологические проявления апоптоза. Апоптоз имеет свои отличительные морфологические признаки, как на светооптическом, так и на ультраструктурном уровне. При окраске гематоксилином и эозином апоптоз определяется в единичных клетках или небольших группах клеток. Апоптотические клетки выглядят как округлые или овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина. Поскольку сжатие клетки и формирование апоптотических телец происходит быстро и также быстро они фагоцитируются, распадаются или выбрасываются в просвет органа, то на гистологических препаратах он обнаруживается в случаях его значительной выраженности. К тому же апоптоз – в отличие от некроза – никогда не сопровождается воспалительной реакцией, что также затрудняет его гистологическое выявление.
Апоптоз – это механизм гибели клеток, который имеет ряд биохимических и морфологических отличий от некроза (рис.45). Наиболее четко морфологические признаки выявляются при электронной микроскопии.
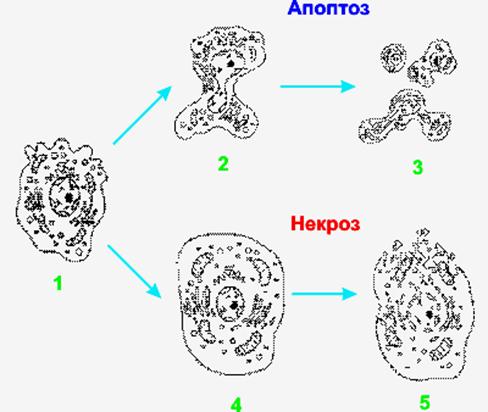
Рис.45. Изменение ультраструктуры клеток животных при некрозе и апоптозе: 1 – нормальная клетка, 2 – апоптотическое сморщивание клетки с образованием пузырчатых выростов, 3 – фрагментация клетки с образованием апоптотических везикул, 4 – набухание клетки при некрозе, 5 – некротическая дезинтеграция клетки.
Для клетки, подвергающейся апоптозу, характерно ее сжатие и конденсация хроматина.
Сжатие клетки. Клетка уменьшается в размерах; цитоплазма уплотняется; органеллы, которые выглядят относительно нормальными, располагаются более компактно. Предполагается, что нарушение формы и объема клетки происходит в результате активации в апоптотических клетках трансглютаминазы. Этот фермент вызывает прогрессивное образование перекрестных связей в цитоплазматических белках, что приводит к формированию своеобразной оболочки под клеточной мембраной, подобно ороговевающим клеткам эпителия.
Конденсация хроматина. Это наиболее характерное проявление апоптоза. Хроматин конденсируется по периферии, под мембраной ядра, при этом образуются четко очерченные плотные массы различной формы и размеров. Ядро же может разрываться на два или несколько фрагментов. Механизм конденсации хроматина изучен достаточно хорошо. Он обусловлен расщеплением ядерной ДНК в местах, связывающих отдельные нуклеосомы, что приводит к развитию большого количества фрагментов, в которых число пар оснований делится на 180-200. При электрофорезе фрагменты дают характерную картину лестницы. Эта картина отличается от таковой при некрозе клеток, где длина фрагментов ДНК варьирует. Фрагментация ДНК в нуклеосомах происходит под действием кальций чувствительной эндонуклеазы. Эндонуклеаза в некоторых клетках находится постоянно (например, в тимоцитах), где она активируется появлением в цитоплазме свободного кальция, а в других клетках синтезируется перед началом апоптоза. Однако еще не установлено, каким образом после расщепления ДНК эндонуклеазой происходит конденсация хроматина. Формирование в цитоплазме полостей и апоптотических телец. В апоптотической клетке первоначально формируются глубокие впячивания поверхности с образованием полостей, что приводит к фрагментации клетки и формированию окруженных мембраной апоптотических телец, состоящих из цитоплазмы и плотно расположенных органелл, с или без фрагментов ядра.
Фагоцитоз апоптотических клеток или телец осуществляется окружающими здоровыми клетками, или паренхиматозными, или макрофагами. Апоптотические тельца быстро разрушаются в лизосомах, а окружающие клетки либо мигрируют, либо делятся, чтобы заполнить освободившееся после гибели клетки пространство.
Фагоцитоз апоптотических телец макрофагами или другими клетками активируется рецепторами на этих клетках: они захватывают и поглощают апоптотические клетки. Один из таких рецепторов на макрофагах – рецептор витронектина, который является β3-интегрином и активирует фагоцитоз апоптотических нейтрофилов.
Апоптоз принимает участие в следующих физиологических и патологических процессах:
· запрограммированном разрушении клеток во время эмбриогенеза (включая имплантацию, органогенез). Несмотря на то, что при эмбриогенезе апоптоз не всегда является отражением “запрограммированной смерти клетки”, это определение апоптоза широко используют различные исследователи,
· гормон-зависимой инволюции органов у взрослых, например, отторжение эндометрия во время менструального цикла, атрезии фолликулов в яичниках в менопаузе и регрессия молочной железы после прекращения лактации,
· удалении некоторых клеток при пролиферации клеточной популяции,
· гибели отдельных клеток в опухолях, в основном при ее регрессии, но также и в активно растущей опухоли,
· гибели клеток иммунной системы, как В-, так и Т-лимфоцитов, после истощения запасов цитокинов, а также гибели аутореактивных Т-клеток при развитии в тимусе,
· патологической атрофии гормон - зависимых органов, например, атрофии предстательной железы после кастрации и истощении лимфоцитов в тимусе при терапии глюкокортикоидами,
· патологической атрофии паренхиматозных органов после обтурации выводных протоков, что наблюдается в поджелудочной и слюнных железах, почках,
· гибели клеток, вызванных действием цитотоксических Т-клеток, например, при отторжении трансплантата и болезни “трансплантат против хозяина”,
· повреждении клеток при некоторых вирусных заболеваниях, например, при вирусном гепатите, когда фрагменты апоптотических клеток обнаруживаются в печени, как тельца Каунсильмана,
· гибели клеток при действии различных повреждающих факторов, которые способны вызвать некроз, но действующих в небольших дозах, например, при действии высокой температуры, ионизирующего излучения, противоопухолевых препаратов.
Молекулярные механизмы апоптоза.Апоптоз – многоэтапный процесс. Первый этап – прием сигнала, предвестника гибели в виде информации, поступающей к клетке извне или возникающей в недрах самой клетки. Сигнал воспринимается рецептором и подвергается анализу.
Далее через рецепторы или их сочетания полученный сигнал последовательно передается молекулам-посредникам (мессенджерам) различного порядка и в конечном итоге достигает ядра, где и происходит включение программы клеточного самоубийства путем активации летальных и/или репрессии антилетальных генов. Однако существование ПКС (программируемая клеточная смерть) в безъядерных системах (цитопластах – клетках, лишенных ядра) показывает, что наличие ядра не является обязательным для реализации процесса.
Применительно к клеткам животных и человека апоптоз в большинстве случаев связан с протеолитической активацией каскада каспаз – семейства эволюционно консервативных цистеиновых протеаз, которые специфически расщепляют белки после остатков аспарагиновой кислоты.
На основе структурной гомологии каспазы подразделяются на подсемейства:
а) каспазы-1 (каспазы 1, 4, 5),
б) каспазы-2 (каспаза-2),
в) каспазы-3 (каспазы 3, 6–10) .
Цистеиновые протеазы, по-видимому, участвуют также в ПКС у растений. Однако апоптоз возможен и без участия каспаз: сверхсинтез белков-промоторов апоптоза Bax и Bak индуцирует ПКС в присутствии ингибиторов каспаз.
В результате действия каспаз происходит:
· активация прокаспаз с образованием каспаз;
· расщепление антиапоптозных белков семейства Bcl-2. Подвергается протеолизу ингибитор ДНКазы, ответственный за фрагментацию ДНК. В нормальных клетках апоптозная ДНКаза CAD (caspase-activated DNase) образует неактивный комплекс с ингибитором CAD, обозначаемым ICAD. При апоптозе ингибитор ICAD с участием каспаз 3 или 7 инактивируется, и свободная CAD, вызывая межнуклеосомальные разрывы хроматина, ведет к образованию фрагментов ДНК с молекулярной массой, кратной молекулярной массе ДНК в нуклеосомных частицах – 180-200 пар нуклеотидов. Апоптоз возможен и без фрагментации ДНК. Обнаружен ядерный белок Acinus (apoptotic chromatin condensation inducer in the nucleus), из которого при комбинированном действии каспазы-3 и неидентифицированной протеазы образуется фрагмент. Этот фрагмент в присутствии дополнительных неядерных факторов вызывает апоптотическую конденсацию хроматина и фрагментацию ядра (кариорексис) без фрагментации ДНК;
· гидролиз белков ламинов, армирующих ядерную мембрану. Это ведет к конденсации хроматина;
· разрушение белков, участвующих в регуляции цитоскелета;
· инактивация и нарушение регуляции белков, участвующих в репарации ДНК, сплайсинге мРНК, репликации ДНК.
Мишенью каспаз является поли(ADP-рибозо)полимераза (ПАРП). Этот фермент участвует в репарации ДНК, катализируя поли(ADP-рибозилирование) белков, связанных с ДНК. Донором ADP-рибозы является NAD+. Активность ПАРП возрастает в 500 раз и более при связывании с участками разрыва ДНК. Апоптотическая гибель клетки сопровождается расщеплением ПАРП каспазами. Чрезмерная активация ПАРП при массированных разрывах ДНК, сильно снижая содержание внутриклеточного NAD+, ведет к подавлению гликолиза и митохондриального дыхания и вызывает гибель клетки по варианту некроза.
Пути реализации программы ПКС (программируемая клеточная смерть).
1. Среди них важное место занимает путь, опосредованный физиологическими индукторами, действие которых реализуется через клеточные рецепторы плазматической мембраны, специально предназначенные для включения программы апоптоза. Этот путь передачи сигнала ПКС схематически можно изобразить следующим образом: индукторы ’ рецепторы ’ адаптеры ’ каспазы первого эшелона ’ регуляторы ’ каспазы второго эшелона. Так, рецептор, обозначаемый Fas, взаимодействуя с соответствующим лигандом (лигандом FasL), трансмембранным белком Т-киллера, активируется и запускает программу смерти клетки, инфицированной вирусом. Тем же путем при взаимодействии с лигандом FasL на поверхности ТН-1-лимфоцитов или с антителом к Fas-рецептору погибают ставшие ненужными выздоровевшему организму В-лимфоциты, продуценты антител, несущие Fas-рецептор. FasL– лиганд, относящийся к многочисленному семейству фактора некроза опухолей TNF. Это семейство гомотримерных лигандов, кроме FasL и TNFa , включает TNFb (лимфотоксин).
Fas – член семейства рецепторов TNF. Все они представлены трансмембранными белками, которые внеклеточными участками взаимодействуют с тримерами лигандов-индукторов. И их взаимодействие запускает в клетке- мишени процесс апоптоза. Тому же способствует и белок перфорин, который выделяется Т-киллерами и образует каналы в мембране клетки-мишени: через эти каналы в клетку проникают протеолитические ферменты гранзимы. Это так называемый инструктивный апоптоз, который абсолютно необходим для нормальной работы иммунной системы млекопитающих. Ключевыми молекулами инструктивного апоптоза являются рецепторы смерти. Лиганды рецепторов смерти представляют собой триммеры. Триммер лиганда связывается с тремя молекулами рецептора, вызывая его триммеризацию. Рецептор имеет цитоплазматический хвост, содержащий домены смерти. От них сигнал поступает к адапторным белкам, которые активируют каспазы – ферменты апоптоза (рис.46.).
Одним из наиболее изученных рецепторов смерти является рецептор Fas, воспринимающий сигнал, который доходит каспазы – 8. Он инициирует апоптоз, активируя другие каспазы – эффекторные. Эти каспазы разрушают клеточные структуры. Таким образом, каспаза – 8 + каспазы–эффекторные это «орудия» апоптоза, а ядерные и цитоплазматические белки – «мишени» каспаз.
Fas и его лиганд (Fas-L) – важные эффекторные молекулы. Ими обладают и используют их цитотоксические иммуноциты в защитной реакции против раковых клеток и клеток, пораженных вирусами.
Чтобы не допустить несвоевременный апоптоз, передачу сигнала через рецепторы смерти контролируют ряд клеточных механизмов:
существуют специальные белки, препятствующие спонтанной агрегации молекул рецептора;
существуют рецепторы-приманки, не содержащие полноценного домена смерти или лишенные цитоплазматического хвоста.
Fas и его лиганд также важны для элиминации активированных иммуноцитов, попавших в иммуннопривилегированные органы – семенники и глаза.
Взаимодействие рецептора и лиганда приводит к образованию кластеров рецепторных молекул и связыванию их внутриклеточных участков с адаптерами.
Адаптер, связавшись с рецептором, вступает во взаимодействие с эффекторами, пока еще неактивными предшественниками протеаз из семейства каспаз первого эшелона (инициирующих каспаз).
Взаимодействие адаптера с рецептором и эффектором осуществляется через гомофильные белок-белковые взаимодействия небольших доменов: DD (death domain – домен смерти), DED (death-effector domain – домен эффектора смерти), CARD (– домен активации и рекрутирования каспазы). Все они имеют сходную структуру, содержат по шесть a-спиральных участков. Домены DD(домен смерти) участвуют во взаимодействии рецептора Fas c адаптером FADD (Fas-associated DD-protein). Домены DED участвуют во взаимодействии адаптера FADD с прокаспазами 8 и 10.
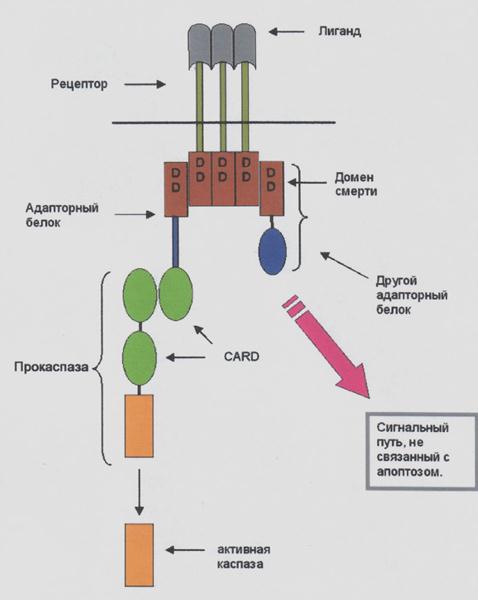
Рис.46. Общая схема передачи сигнала через рецепторы смерти.
Наиболее подробно охарактеризована прокаспаза-8, рекрутируемая рецептором Fas через адаптeр FADD. Образуются агрегаты FasL – Fas – FADD – прокаспаза-8. Подобные агрегаты, в которых происходит активация каспаз, названы апоптосомами, апоптозными шаперонами, или сигнальными комплексами, индуцирующими смерть.
Прокаспазы обладают незначительной протеолитической активностью, составляющей 1–2% активности зрелой каспазы. Будучи в мономерной форме, прокаспазы, концентрация которых в клетке ничтожна, находятся в латентном состоянии. Предполагается, что пространственное сближение молекул прокaспаз при их агрегации ведет к образованию активных каспаз через механизм протеолитического само- и перекрестного расщепления (ауто- или транс-процессинга). В результате от прокаспазы (молекулярная масса 30–50 кДа) отделяется регуляторный N-концевой домен (продомен), а оставшаяся часть молекулы разделяется на большую (~20 кДа) и малую (~10 кДа) субъединицы. Затем происходит ассоциация большой и малой субъединиц. Два гетеродимера образуют тетрамер с двумя каталитическими участками, действующими независимо друг от друга. Таким образом, прокаспаза-8 активируется и высвобождается в цитоплазму в виде каспазы-8. Существуют другие пути активации каспазы-8 – с участием рецепторов TNFR1 и DR3.
На этапе активации каспаз первого эшелона жизнь клетки еще можно сохранить. Существуют регуляторы, которые блокируют или, напротив, усиливают разрушительное действие каспаз первого эшелона. К ним относятся белки Bcl-2 (ингибиторы апоптоза: A1, Bcl-2, Bcl-W, Bcl-XL, Brag-1, Mcl-1 и NR13) и Bax (промоторы апоптоза: Bad, Bak, Bax, Bcl-XS, Bid, Bik, Bim, Hrk, Mtd). Эти белки эволюционно консервативны: гомолог Bcl-2 обнаружен даже у губок, у которых апоптоз необходим для морфогенеза.
Каспаза-8 активирует каспазу второго эшелона (эффекторную каспазу): путем протеолиза из прокаспазы-3 образуется каспаза-3, после чего процесс, запущенный программой смерти, оказывается необратимым.
Каспаза-3 способна в дальнейшем к самостоятельной активации (автокатализу или автопроцессингу), активирует ряд других протеаз семейства каспаз, активирует фактор фрагментации ДНК, ведет к необратимому распаду ДНК на нуклеосомальные фрагменты. Так запускается каскад протеолитических ферментов, осуществляющих апоптоз.
2. Второй путь реализации программы ПКС связан с митохондриальным цитохромом c. В клетках, подвергшихся воздействию индуктора апоптоза, резко снижается мембранный потенциал (Dy) митохондрий. Падение Dy обусловлено увеличением проницаемости внутренней мембраны митохондрий вследствие образования гигантских пор. Существует много факторов, вызывающих раскрытие пор. К ним относятся истощение клеток восстановленным глутатионом, NAD(P)H, ATP и ADP, образование активных форм кислорода, разобщение окислительного фосфорелирования протонофорными соединениями, увеличение содержания Ca2+ в цитоплазме. Образование пор в митохондриях можно вызвать церамидом, NO, каспазами, амфипатическими пептидами, жирными кислотами. Поры имеют диаметр 2,9 нм, позволяющий пересекать мембрану веществам с молекулярной массой 1,5 кДа и ниже. Следствием раскрытия пор является набухание митохондриального матрикса, разрыв наружной мембраны митохондрий и высвобождение растворимых белков межмембранного объема. Среди этих белков – ряд апоптогенных факторов: цитохром с, прокаспазы 2, 3 и 9, белок AIF (apoptosis inducing factor), представляющий собой флавопротеин с молекулярной массой 57 кДа.
Образование гигантских пор не является единственным механизмом выхода межмембранных белков митохондрий в цитоплазму. Предполагается, что разрыв наружной мембраны митохондрий может быть вызван гиперполяризацией внутренней мембраны. Возможен и альтернативный механизм, без разрыва мембраны, – раскрытие гигантского белкового канала в самой наружной мембране, способного пропускать цитохром с и другие белки из межмембранного пространства.
Высвобождаемый из митохондрий цитохром с вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) участвует в активации каспазы-9. APAF-1 – белок с молекулярной массой 130 кДа, содержащий CARD-домен (caspase activation and recruitment domain) образует комплекс с прокаспазой-9 в присутствии цитохрома с и dATP или АТР. Из этих субъединиц собираются жесткие, симметричные структуры, наподобие веера или пропеллера. APAF-1 играет роль арматуры, на которой происходит аутокаталитический процессинг каспазы-9. Предполагается, что в результате зависимого от гидролиза dATP (или АТР) конформационного изменения APAF-1 приобретает способность связывать цитохром с (рис.47). Связав цитохром с, APAF-1 претерпевает дальнейшее конформационное изменение, способствующее его олигомеризации и открывающее доступ CARD-домена APAF-1 для прокаспазы-9, которая тоже содержит CARD-домен. Так образуется конструкция, называемая также апоптосомой, с молекулярной массой > 1,3 млн дальтон, в составе которой – не менее 8 субъединиц APAF-1. Благодаря гомофильному CARD-CARD-взаимодействию с APAF-1 в эквимолярном соотношении связывается прокаспаза-9, а затем прокаспаза-9 связывает прокаспазу-3. Пространственное сближение молекул прокаспазы-9 на мультимерной арматуре из APAF-1-цитохром-с-комплексов, по-видимому, приводит к межмолекулярному протеолитическому процессингу прокаспазы-9 с образованием активной каспазы-9. Зрелая каспаза-9 затем расщепляет и активирует прокаспазу-3.
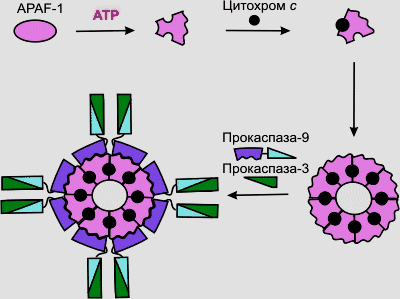
Рис.47. Флавопротеин - митохондриальный эффектор апоптоза у животных
Флавопротеин AIF, будучи добавленным, к изолированным ядрам из клеток HeLa, вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям печени крыс (высвобождение цитохрома с и каспазы) AIF является митохондриальным эффектором ПКС, действующим независимо от каспаз (рис.47).
Кроме рассмотренных компонентов, при нарушении наружной мембраны митохондрий из межмембранного объема выделяется термолабильный фактор, вызывающий необратимое превращение ксантиндегидрогеназы в ксантиноксидазу. Ксантиндегидрогеназа катализирует зависимое от NAD+ окисление ксантина до гипоксантина и последующее окисление гипоксантина до мочевой кислоты. Ксантиноксидаза катализирует те же реакции, но не с NAD+, а с О2 в качестве акцептора электронов. При этом образуются О2A, Н2О2, а из них – и другие активные формы кислорода (АФК), которые разрушают митохондрии и являются мощными индукторами апоптоза. Механизмы образования АФК, конечно, не ограничиваются ксантиноксидазной реакцией. Главным источником АФК в клетках являются митохондрии. Резкое увеличение АФК происходит при возрастании мембранного потенциала в митохондриях, когда снижено потребление ATP и скорость дыхания лимитируется ADP . Цитоплазматическая мембрана макрофагов и нейтрофилов содержит О2A – генерирующую NADPH-оксидазу.
В зависимости от пути, по которому осуществляется активация каспаз, различают разные типы клеток. Клетки типа I (в частности, линия лимфобластоидных В-клеток SKW и T-клетки линии Н9) подвергаются ПКС по пути, зависимому от апоптозных рецепторов плазматической мембраны без участия митохондриальных белков. Клетки типа II (например, линии Т-клеток Jurkat и СЕМ) погибают по пути апоптоза, зависимому от митохондриального цитохрома с. ПКС, вызванная химиотерапевтическими соединениями, УФ- или і-облучением, по-видимому, напрямую связана с апоптозной функцией митохондрий.
Некоторые клетки, например, клетки эмбриональной нервной системы, включают механизмы апоптоза, если они испытывают дефицит апоптозподавляющих сигналов (называемых также факторами выживания) от других клеток. Физиологический смысл процесса – в элиминации избыточных нервных клеток, конкурирующих за ограниченный фонд факторов выживания. Эпителиальные клетки при отделении от внеклеточного матрикса, вырабатывающего факторы выживания, тоже обречены на ПКС. Факторы выживания связываются соответствующими цитоплазматическими рецепторами, активируя синтез подавляющих апоптоз агентов и блокируя стимуляторы апоптоза. Некоторые вещества (например, стероидные гормоны) оказывают дифференцированный эффект на различные типы клеток – предотвращают апоптоз одних типов клеток и индуцируют его у других. Так, при наличии во внеклеточном матриксе факторов роста PDGF (platelet-derived growth factor – тромбоцитарный фактор роста) или NGF (nerve growth factor – фактор роста нервов) и цитокина интерлейкина-3 (IL-3) проапоптозный белок Bad не активен.Факторы роста, связавшись со своим рецептором на плазматической мембране, вызывают активацию цитозольной протеинкиназы В, и катализирующей фосфорилирование Bad по Ser-136. IL-3 тоже связывается со своим рецептором на плазматической мембране и акти
– Конец работы –
Эта тема принадлежит разделу:
ПАВЛИЧЕНКО В. И АБРАМОВ А. В.
На сайте allrefs.net читайте: ПАВЛИЧЕНКО В. И.
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Регуляция клеточного цикла. Апоптоз. Онкогенетика.
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.038 сек.
Новости и инфо для студентов