Реакции карбоновых кислот с нуклеофильными реагентами
1.1 Oбразование солей с металлами:
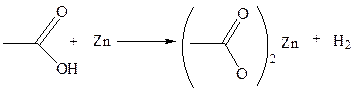
1.2 Взаимодействие с оксидами металлов:
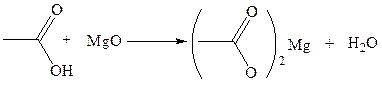
1.3 Взаимодействие с основаниями:
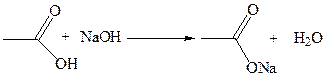
1.4 Взаимодействие с аммиаком:
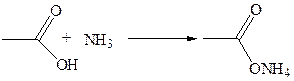
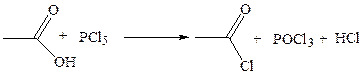
1.5 Образование галогенангидридов:
1.6 Реакции с Н-нуклеофилами (комплексными гидридами). Комплексные гидриды привзаимодействии с карбоновыми кислотами выделяют водород и образуют карбоксилаты, которые в избытке реагента и при более жестких условиях восстанавливаются до первичных спиртов:
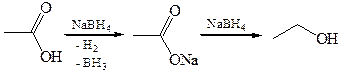
1.7 Реакции с N-нуклеофилами. N-Hуклеофилы (аммиак, амины, гидразин и др.) при взаимодействии с карбоновыми кислотами, как правило, образуют аммониевые карбоксилаты и только при повышенных температурах происходит присоединение к карбонильной группе и получаются амиды:
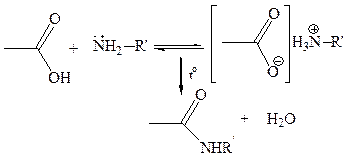
1.8 Реакции с О-нуклеофилами (реакция этерификации по Фишеру). O-Hуклеофилы нейтрального характера, например, спирты, реагируют и образуют сложные эфиры (часть 2, главы 3.1, 6.3.4):
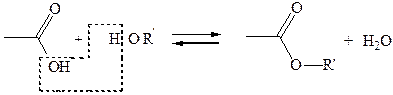
2. Реакции с электрофильными реагентами. Карбоновые кислоты, как очень слабые нуклеофилы, взаимодействуют только с особенно сильными реагентами, присоединение происходит по кислородному атому. Для осуществления реакции с более слабыми электрофильными реагентами необходимокарбоксильную группу активировать, что осуществляется превращением ее в карбоксилат-ион. Тогда можно осуществить реакции алкилирования, ацилирования и др.
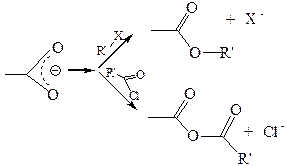
3. Реакции нуклеофильного замещения у карбоновых кислот, идут по электрофильному центру ацила и имеют одинаковый в главных стадиях механизм, который называется тетраэдрическим, характерный для всех классов функциональных производных карбоновых кислот. Реакция заключается в «передаче» ацила от субстрата к нуклеофильному реагенту, поэтому процесс называется реакцией ацилирования, а субстрат – ацилирующим агентом. По реагенту реакции ацилирования различают как гидролиз (нуклеофильный реагент – вода), алкоголиз (спирт), аммонолиз (аммиак), ацидолиз (кислота) и некоторые другие. Скорость реакций ацилирования при равных внешних условиях определяется природой субстрата (ацилирующего агента) и нуклеофила. Механизм реакций ацилирования можно представить следующей общей схемой:
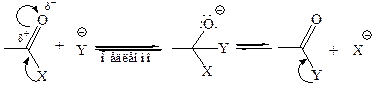
Таким образом, реакции ацилирования протекают в две стадии:
· присоединение к карбонильному атому углерода нуклеофила за счет собственной пары электронов, сопровождающееся гетеролизом π-связи, эта стадия протекает по AN механизму и является лимитирующей;
· отщепление уходящей группы, протекающее очень легко, так как в результате происходит восстановление сопряженной системы.
По своему результату реакции ацилирования могут быть отнесены к реакциям SN2. От обычных SN2 реакций у алкилгалогенидов и спиртов, протекающих у насыщенного реакционного центра, они отличаются тем, что являются двухстадийными и протекают более легко. Большая скорость реакций ацилирования в сравнении со скоростью SN реакций у насыщенного атома углерода объясняется следующим образом:
· реакционный центр в ацильной группе более положителен;
· меньше стерические препятствия при атаке нуклеофила вследствие плоского строения реакционного центра в исходном субстрате и наличия в промежуточном аддукте у реакционного центра только четырех заместителей.
Скорость SN реакций может уменьшаться при увеличении размера нуклеофила. Поскольку лимитирующей стадией реакции ацилирования является AN стадия, то реакционную способность ацилирующего средства определяет величина δ+ на карбонильном углероде. Следовательно, чем выше электроноакцепторное действие заместителя X, тем больше величина δ+ на реакционном центре и тем выше скорость ацилирования. В силу этого ацилирующие средства можно расположить в порядке уменьшения их ацилирующей способности следующим образом:
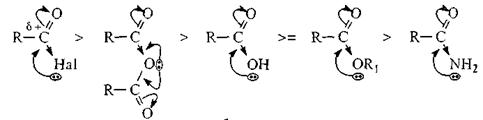
Соли карбоновых кислот ацилирующей способностью не обладают, так как в карбоксилатанионе на карбонильном углероде вследствие делокализации электронов слишком низок δ+ и атом углерода теряет способность подвергаться атаке нуклеофила.
Как видно из механизма реакции, от промежуточного аддукта может отщепляться не только группа X–, но и группа Y+, т. е. может идти и обратная реакция. Поэтому многие реакции ацилирования – равновесные процессы. Положение равновесия зависит от соотношения скоростей прямой и обратной реакций, и в результате превращения всегда будет образовываться менее реакционноспособное вещество. Поэтому активность ацилирующего средства влияет не только на скорость реакции ацилирования, но и на положение равновесия. В случае использования сильных ацилирующих средств (хлорангидридов и ангидридов) равновесие сильно сдвинуто вправо и реакции можно считать необратимыми. Если же различие в реакционной способности субстрата и продукта реакции невелико, то резкого смещения равновесия реакции не наблюдается, т. е. реакция обратима. Так, сильные ацилирующие агенты хлорангидриды и ангидриды легко реагируют со спиртами и аммиаком с образованием сложных эфиров и амидов. Сложные эфиры реагируют с аммиаком с образованием амидов. Однако обратные реакции – образование сложных эфиров из амидов, – хотя и возможны, но их довольно трудно осуществить.
В ряде случаев ацилирование проводят в условиях либо кислотного, либо основного катализа. Поскольку реакция начинается с нуклеофильного присоединения к карбонильной группе, роль катализатора при ацилировании аналогична роли катализатора в AN реакциях альдегидов и кетонов.
4. Реакции α-водородного атома
4.1 Галогенирование кислот. Подобно альдегидам и кетонам у карбоновых кислот водород у α-углеродного атома приобретает повышенную подвижность. Однако поскольку величина δ+ на атоме углерода карбоксильной группы понижена за счет +М эффекта, то влияние карбоксила на α-углеродный атом значительно меньше, чем у альдегидов и кетонов.
Благодаря подвижности атомов водорода связи Сα–Н карбоновые кислоты могут подвергаться свободнорадикальному галогенированию. Однако в этом случае вследствие высокой активности и малой избирательности атомов хлора галогенирование может происходить и по другим положениям цепи:
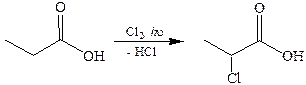
5. Реакция Геля–Фольгарда–Зелинского. Осуществляют галогеном в присутствии небольших количеств красного фосфора:
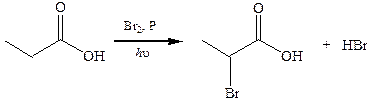
6. Реакции декарбоксилирования. Одноосновные кислоты довольно устойчивы к нагреванию. Однако если их нагревать при температуре выше 300 °С с MnO2 или ThO2, то происходит декарбоксилирование с образованием альдегидов или кетонов. Если в углеводородном остатке кислоты имеются сильные электроноакцепторные группы, то возможно довольно легкое отщепление СО2 при температурах 100–150 °С.
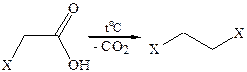
где Х= –NO2,–COOH, –C=O
6.1 Реакция Кольбе (электролиз солей карбоновых кислот, часть 1, глава 8.1).
6.2 Образование оксосоединений. При пиролизе кальциевых или бариевых солей карбоновых кислот образуются оксосоединения:
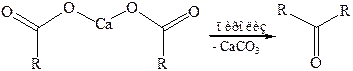
|
6.3. Образование галогеналканов (реакция Хунсдиккера). Декарбокси-лирование происходит также при взаимодействии серебряных или ртутных солей карбоновых кислот с йодом или бромом. Эта реакция лучше всего проходит с первичными кислотами:
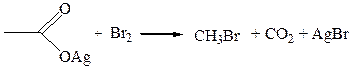
|
Предполагают, что промежуточным продуктом является гипогалогенит, который распадается на радикалы:
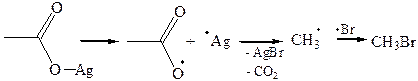
|
6.4. Пиролиз карбоновых кислот (образование кетенов) протекает при 700–800 °С с образованием кетенов:
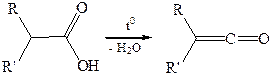
|
6.5. Дегидратация карбоновых кислот протекает при высоких температурах с образованием кетенов:
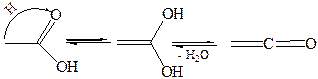
|
6.6 Реакция Дюма (часть 1, глава 8.1)
Отдельные представители. Муравьиная кислота – бесцветная едкая жидкость с острым запахом, смешивающаяся с водой. Впервые выделена в XVII веке из красных муравьев перегонкой с водяным паром. В природе встречается в свободном состоянии в крапиве.
Муравьиная кислота по своему строению и свойствам отличается от остальных членов гомологического ряда предельных монокарбоновых кислот. В отличие от других карбоновых кислот в молекуле муравьиной кислоты функциональная карбоксильная группа связана не с углеводородным радикалом, а с атомом водорода. По этой причине муравьиная кислота является наиболее сильной кислотой по сравнению с другими членами своего гомологического ряда.
В молекуле муравьиной кислоты можно выделить и альдегидную группу, поэтому кислота вступает в реакции, характерные как для кислот, так и для альдегидов. В частности, как и альдегиды, НСООН проявляет восстановительные свойства.
Муравьиная кислота широко используется в органическом синтезе, например, для получения формамида, диметилформамида, щавелевой кислоты. Муравьиная кислота обладает бактерицидным действием, поэтому используется для консервирования и для дезинфекции емкостей для пива и вина. Формиат алюминия используют для пропитки текстильных изделий. Применяют муравьиную кислоту и при крашении тканей.
Уксусная кислота представляет собой бесцветную жидкость с острым запахом и кислым вкусом, неограниченно смешивающуюся с водой. Безводную уксусную кислоту называют «ледяной», так как при 16 °С она замерзает и образует кристаллы, подобные льду. Обычная уксусная кислота, содержащая 2–3 % воды, замерзает при температуре ниже 13 °С.
Разбавленные водные растворы уксусной кислоты образуются при брожении вина. При перегонке водных растворов получают приблизительно 80 % кислоту («уксусную эссенцию»), которую применяют для пищевых целей.
Уксусную кислоту используют в качестве растворителя и как исходное вещество для синтеза производных уксусной кислоты (ацетилхлорида, ацетангидрида, амидов, сложных эфиров). Ацетаты применяют в текстильной промышленности в качестве протравителей и в синтезе как основные катализаторы, а также их используют для борьбы с вредителями растений, 3–6 % растворы (столовый уксус) используют как вкусовую приправу и консервант. Эфиры уксусной кислоты применяют в качестве растворителей лаков и красок. Многие эфиры и амиды уксусной кислоты используются в медицине в качестве лекарственных средств.
Пальмитиновая кислота представляет собой бесцветное кристаллическое вещество со слабым запахом стеарина, в воде не растворяется, широко распространена в природе, в виде сложных эфиров с глицерином входит в состав жиров. Получают пальмитиновую кислоту обработкой жиров щелочью (гидролиз, омыление). При этом образуются соли (пальмитаты), после подкисления, которых осаждается сама кислота. Пальмитиновая кислота и ее производные используются в качестве ПАВ (моющих средств и др.), а ее натриевая соль называется мылом.
Стеариновая кислота – бесцветное кристаллическое вещество со слабым запахом стеарина. Ее эфиры с глицерином входят в состав жиров.
Получают стеариновую кислоту омылением жиров, обычно образуется смесь стеариновой и пальмитиновой кислот. Стеариновую кислоту в смеси с пальмитиновой используют в производстве свечей, их натриевые соли являются обыкновенным мылом. В органическом синтезе стеариновую кислоту используют для получения других поверхностно-активных веществ. Производные пальмитиновой и стеариновой кислот принадлежат к важным природным веществам – липидам.
Наиболее важные источники алифатических карбоновых кислот – животные и растительные жиры, которых можно получить кислоты с неразветвленной цепью и с четным числом атомов углерода, начиная от шести и до восемнадцати атомов углерода.