рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Физика
- /
- Синдром Ваарденбурга
Реферат Курсовая Конспект
Синдром Ваарденбурга
Синдром Ваарденбурга - раздел Физика, Генетические нарушения и их проявление у лиц с особенностями психофизического развития Синдром Ваарденбургаимеет Следующие Клинические Признаки: Телекант (Латеральн...
Синдром Ваарденбургаимеет следующие клинические признаки: телекант (латеральное смещение внутреннего угла глаза), гетерохромия радужки, седая прядь надо лбом и врожденная глухота.
Телекант в сочетании с широкой и приподнятой спинкой носа и сросшимися бровями создает весьма своеобразный облик пораженных – «греческий профиль». Очень характерны сросшиеся брови. Радужки либо различно окрашены (один глаз голубой, другой – карий), либо имеется сектор иного цвета в одной из радужек. У больных очень редко можно выявить весь набор типичных признаков: каждый симптом имеет свою степень экспрессивности. С наибольшим постоянством проявляется телекант – у 99% носителей гена, широкая спинка носа – 75%, сросшиеся брови – у 45%, гетерохромия радужки – у 25%, седая прядь или ранняя седина – у 17% наблюдавшихся носителей гена.
Кроме указанных признаков, у больных иногда есть участки гипер– и депигментации на коже, пигментные изменения глазного дна. Седая прядь бывает уже у новорожденного, но затем эти депигментированные волоски часто исчезают. Нос часто имеет не только приподнятую спинку, но и гипоплазию крыльев.
Таблица 5
Генные болезни, соответствующие определенным типам наследования
| Тип наследования | Заболевание | Локализация мутантного гена | Критерии наследования |
| аутосомно–доминантный | Синдром Ваарденбурга | 2q37 (атрофия кортиева органа, врожденная глухота) | · Проявление признака у гетерозиготных носителей гена. · При анализе родословной признак выявляется в каждом поколении. · Пенетрантность патологических проявлений почти всегда ниже 100%. · Различная выраженность клинических проявлений не только между разными семьями, но и внутри каждой семьи. · Клинические признаки могут появиться не сразу после рождения, а спустя много лет. · Здоровые члены семьи не могут иметь больных детей. |
| Синдром Марфана | 15q21 (порок развития соединительной ткани) | ||
| Синдром Реклингхау-зена (нейрофибро-матоз) | 22q12 (супрессор опухолевого роста) | ||
| аутосомно–рецессивный | Фенил-кетонурия (ФКУ) | 12q22 (нет синтеза фенилаланин-гидроксилазы) | · Мутантный ген проявляется только у гомозигот по рецессивному гену. · Если родители гетерозиготны, то вероятность рождения больного ребенка составляет 25%. · При анализе родословной мутантный ген проявляется не в каждом поколении. · Вероятность проявления мутантного гена возрастает в родственных браках. · Частота проявления мутантного гена у лиц женского и мужского пола одинакова. |
| Гомоцисти-нурия | 21q22 (нет синтеза цистатионин-синтетаза) | ||
| Галактоземия | 9р13 (нет синтеза галактозо-1-фосфат-уридилтрансферазы | ||
| Синдром Ушера | 14q | ||
| сцепленный с полом (рецессивный, сцепленный с X-хромосомой) | Синдром Мартина – Белла (ломкой Х–хромосомы) | Хq27 (? порок развития соединительной ткани) | · Мутантный ген (рецессивный) проявляется преимущественно у лиц мужского пола. · Если отец болен, мать здорова (фенотип, генотип), то все дочери будут гетерозиготными носительницами. Половая Х–хромосома от отца передается только дочерям. · Если отец здоров, мать фенотипически здорова (т.е. она носительница мутантного гена), то вероятность рождения больных сыновей составит 50%. · Если мутантный ген, локализованный в Х–хромосоме, является доминантным, то он проявляется и у мужчин, и у женщин. Частота заболевания женщин в популяции в 2 раза больше. |
| Синдром Дюшена (псевдогипертрофическая мышечная дистрофия | Хр16 (мутация гена дистрофина, кодирующего структурный белок сарколеммы). |
Патология конечностей включает такие аномалии, как гипоплазия кистей и мышц, ограничение подвижности локтевых, лучезапястных и межфаланговых суставов, слияние отдельных костей запястья и плюсны.
Снижение слуха при этом заболевании врожденное, воспринимающего типа, связанное с атрофией преддверно–улиткового органа (кортиев орган). Глухота вызвана нарушениями спирального (кортиева) органа с атрофическими изменениями в спинальном узле и слуховом нерве.
Синдром Ваарденбурга встречается с частотой 1:4000. Среди детей с врожденной глухотой составляет 3%. Синдром определяется аутосомно-доминантным геном с неполной пенетрантностью и варьирующейся экспрессивностью. Ген локализован на хромосоме 2q37.
При лечении в некоторых случаях показана косметическая хирургия телеканта. Лечение глухоты неэффективно.
Синдром Марфана
Синдром Марфанаобусловлен пороком развития соединительной ткани и характеризуется поражением, в первую очередь, опорно–двигательного аппарата. Больные, как правило, имеют высокий рост, характеризуются диспропорцией в росте туловища и конечностей, кисти и стопы у них длинные с тонкими «паукообразными пальцами», грудная клетка килевидной или воронкообразной формы. Для синдрома характерен кифоз, сколиоз, широкие межреберные промежутки, тонкие и длинные ребра, которые имеют отвесное направление; «птичье» выражение лица (узкий череп, подбородок срезан или выступает, близко посаженные глаза, ушные раковины тонкие и малоэластичные), перерастяженность сухожилий и суставов, слабость связок, мышечная гипотония, недоразвитие подкожной клетчатки.
Наблюдается патология глаз, а именно, миопия, голубые склеры, частичный или полный подвывих хрусталика, колобома радужной оболочки. Часты нарушения внутренних органов: сердечно–сосудистой системы (пороки сердца, крупных сосудов, расслаивающаяся аневризма аорты, аномалия расположения сосудов), уменьшение числа долей легких.
Умственное развитие при этом заболевании обычно не страдает.
Синдром Марфана встречается в общей популяции с частотой 1: 10000. Синдром определяется доминантным геном с различной экспрессивностью. Ген локализован на хромосоме 15q21. Нормальный рецессивный аллель этого гена кодирует белок фибриллин, участвующий в формировании волокон коллагена из проколлагена. Мутации гена приводят к недоразвитию (или к разрушению) значительной части волокон коллагена, являющегося важнейшим компонентом соединительной ткани.
Рентгенологически определяются остеопороз метафизарных отделов костной ткани, истончение кортикального слоя, шпорообразные пяточные кости. В сыворотке крови повышен уровень кислых мукосахаридов, снижено содержание серомукоида. В моче повышено содержание мукополисахаридов (хондроитинсульфат, кератосульфат), гидроксипропилина.
Дифференциальный диагноз с синдромом Стиклера, гигантизмом, акромегалией. Лечение симптоматическое.
Синдром Реклингхаузена
Синдром Реклингхаузена(нейрофиброматоз Реклингхаузена)одно из самых частых моногенных наследственных заболеваний: его популяционная частота составляет 1:3000 новорожденных. В контингенте умственно отсталых детей нейрофиброматоз встречается на порядок чаще; среди учащихся вспомогательных школ–интернатов с частотой 1:260.
В зависимости от распространенности и локализации новообразований заболевание подразделяется на периферическую и центральную форму – соответственно, нейрофиброматоз-1 (NF1) нейрофиброматоз-2 (NF2).
Ген NF1 локализован на хромосоме 17q11 и состоит из 60 экзонов. Ген NF1 кодирует белок нейрофибромин, являющийся супрессором опухолевого роста. У больных нейрофиброматозом-1 в гене NF1 выявлено свыше 200 мутаций.
Основным клиническим признаком заболевания в детском возрасте являются множественные «кофейные» пятна на коже, иногда они имеются уже при рождении, но чаще появляются несколько позднее, как правило в первом десятилетии жизни. Они постепенно увеличиваются в числе и размерах. Обычно их форма овальная, они расположены на различных частях тела, но чаще на груди, спине, животе. Размеры пятен различны – от точечных до нескольких сантиметров в диаметре. Патогномоничны для заболевания высыпания мелких кофейных пятен, похожих на веснушки, в подмышечной ямке.
На коже можно отметить и другие изменения: сосудистые пятна, участки депигментации, гипертрихоз, очаговое поседение волос. С возрастом на коже у больных появляются весьма характерные мягкие на ощупь светлые опухоли, имеющие форму папиллом или более плоские. Эти высыпания при надавливании как бы проваливаются в кожу – симптом «кнопки звонка». Их число сильно варьирует – от единичных до нескольких сотен. Данный признак отмечается обычно только у подростков и взрослых, реже у детей старшего возраста, у маленьких детей – не обнаружен.
Помимо кожных высыпаний, встречаются подкожные опухоли, так называемые плексиформные невромы. Они обычно округлые («бусинки»), диаметром 1–2 см, редко крупнее, подвижные, не прикреплены к коже, лежат по ходу нервных стволов. Иногда подкожных опухолей много, в других случаях удается обнаружить не более 1–2 узелков. Скорость роста опухолей очень различна. Обычно они не изменяются несколько лет, а потом вдруг могут начать быстро расти. Как правило, опухоли не нарушают функции периферических нервов, но при сильном сдавлении нервного ствола могут вызвать боль, парезы и параличи.
Расположенные по ходу черепных нервов невромы могут нарушать их функцию, что нередко сопровождается снижением слуха или зрения и другими симптомами – нейрофиброматоз-2 (NF2). Ген NF2 локализован на хромосоме 22q12. Продукт гена состоит из 587 аминокислот - белок (мерлин). Является ингибитором опухолевого роста.
Можно отметить некоторое сходство облика детей, страдающих нейрофиброматозом: голова, как правило, крупная, черты лица грубоватые, несколько акромегалоидные, кисти рук и стопы большие, широкие, шея короткая. Очень часто грудная клетка деформирована – куриная грудь, вдавленная грудина. В более старшем возрасте у мальчиков отмечается некоторая евнухоидность: высокий таз, длинные ноги, задержка появления вторичных половых признаков. Нередко у детей имеются и врожденные пороки развития: вывих тазобедренного сустава, пороки сердца. При опухолях в полости черепа развивается самая различная симптоматика в зависимости от их локализации и темпов роста: деменция, эпилепсия, афазия и т.п.
Специфического лечения нет. В ряде случаев опухоли удаляют хирургическим путем. Патогенез заболевания связан с опухолевыми разрастаниями, поэтому стимулирующие препараты детям назначать не рекомендуется. Проводится лечение гидроцефалии.
Фенилкетонурия (ФКУ)
Фенилкетонурия – наследственное заболевание обмена, характеризующееся поражением ЦНС и прогрессирующим, особенно в первые 2–3 года жизни, слабоумием. Фенотипически здоровые родители больного ребенка являются гетерозиготными носителями мутантного гена. Частота заболевания в Европе в среднем составляет 1:10 000 новорожденных, распространенность носителей гена в популяции 1:50. ФКУ наблюдается примерно у 1% умственно отсталых лиц.
Заболевание обусловлено мутацией гена, контролирующего синтез фермента фенилаланингидроксилазы, который обеспечивает превращение поступающего в организм с пищей фенилаланина в тирозин (рис. 21).
Нарушение последнего процесса приводит к резкому повышению содержания фенилаланина в сыворотке крови и спинномозговой жидкости, при этом отмечают дефицит тирозина, что определяет недостаточный синтез катехоламинов, гормона щитовидной железы и меланина, при недостаточном количестве которого наблюдается слабая пигментация кожи и волос. При ФКУ нарушается также обмен триптофана и синтез серотонина, что губительно действует на нормальное функционирование нервной системы. Ген РАН локализован на хромосоме 12q22.
Дети с ФКУ рождаются с полноценным головным мозгом, так как биохимические процессы плода осуществляются за счет процессов в организме матери. Возникающие после рождения биохимические нарушения оказывают токсическое воздействие на нервную систему, в результате чего нарушаются процессы миелинизации, развитие и рост мозга.
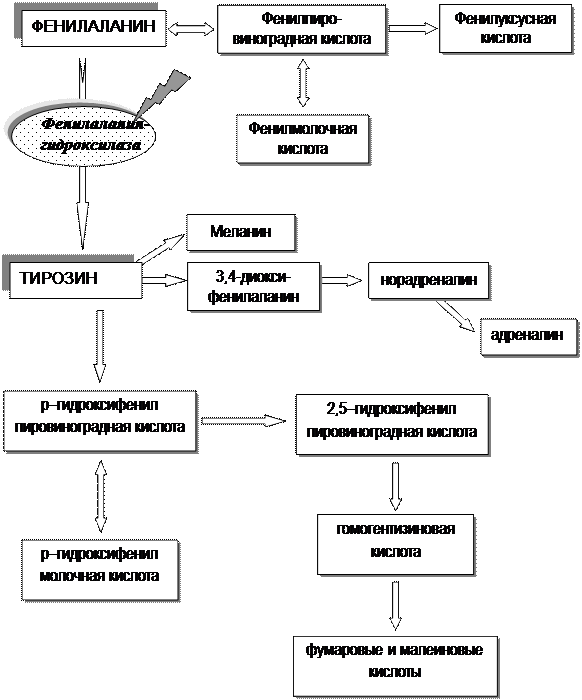
|
| Рис.21. Схема обмена фенилаланина и тирозина при фенилкетонурии |
Нарастание интеллектуального дефекта сочетается с отставанием в физическом развитии, часто с признаками умеренной микроцефалии. Характерен внешний вид больных (блондины со светлой кожей и голубыми глазами) и отдельные диспластические признаки (высокое небо, эпикант, деформация ушных раковин).
При этом отмечают следующие неврологические нарушения: мышечную гипертонию, повышение сухожильных рефлексов, гиперкинезы, тремор пальцев рук, атаксию, нарушения черепно–мозговой иннервации. В более редких случаях имеет место мышечная гипотония; судорожный синдром наблюдается у 20–50% больных.
Уровень интеллектуального развития колеблется от нормы до глубокой идиотии. Прогредиентность динамики слабоумия наиболее выражена в первые 2–3 года жизни. Больные отличаются инертностью, недостаточной целенаправленностью с характерными нарушениями внимания, памяти, недоразвитием гностических функций и пространственных представлений.
Отмечается также выраженное недоразвитие речи и нарушения звукопроизношения. Нарушения речи обычно сопоставимы с глубиной интеллектуального дефекта.
Гомоцистинурия
Гомоцистинурияобусловлена отсутствием или снижением активности фермента цистатионинсинтетазы, необходимого для синтеза цистатионина из гомоцистеина и серина (рис.22). Ген локализован на хромосоме 21q22.
Дефект генетически гетерогенен. Существуют две формы, различающиеся по отношению к витамину В6: пиридоксинзависимая и пиридоксинрезистентная. Описаны случаи заболевания, вызванные дефицитом других ферментов. Частота заболевания среди новорожденных колеблется от 1:80 000 до 1:180 000. Среди умственно-отсталых частота гомоцистинурии достигает 0.3%. В контингенте умственно отсталых с дефектами зрения – 2.6%.
| ||
| Рис.22. Обмен метионина при гомоцистинурии |
Клиническая картина полиморфна, но вместе с тем наиболее типичным симптомокомплексом считается сочетание умственной отсталости с дефектами зрения (эктопия хрусталика, катаракта, миопия) и костной системы (удлинение трубчатых костей при укороченном туловище, деформация суставов, деформация стоп, крыловидные лопатки). Внешними, наиболее выраженными признаками являются мягкие светлые волосы, голубые радужки, диспропорциональность телосложения с укорочением туловища и удлинением конечностей в сочетании со многими стигмами дисэмбриогенеза (воронкообразная грудная клетка, остеопороз костей и др.). Поражение соединительной ткани, механизм которого еще не ясен, определяет сходство гомоцистинурии с болезнью Марфана. Существует предположение о патогенетической роли в патогенезе гомоцистинурии дефицита меди.
Нервно–психические нарушения при этом заболевании отмечаются в 75% случаев. Описаны легкие (пограничные) и глубокие формы умственной отсталости с инертностью нервных процессов, недостаточной критичностью, расстройством речи. В ряде случаев отмечены двигательные нарушения в виде параличей и парезов. Нарушения речи включают общее недоразвитие, косноязычие, дизартрию.
Биохимическая диагностика направлена на качественное определение цистина и гомоцистина в моче, а также количественное определение метионина и гомоцистина в плазме на аминокислотном анализаторе. С целью ПД определяется активность цистатионинсинтетазы в культуре амниотических клеток. Лечение заключается в диете, бедной метионином. При пиридоксинзависимой форме заболевания эффективна терапия большими дозами витамина В6.
– Конец работы –
Эта тема принадлежит разделу:
Генетические нарушения и их проявление у лиц с особенностями психофизического развития
Белорусский государственный педагогический университет... имени Максима Танка...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Синдром Ваарденбурга
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.022 сек.
Новости и инфо для студентов