
Расчет энергетических величин и выражения первого закона термодинамики для предельных процессов идеального газа
Расчет энергетических величин и выражения первого закона термодинамики для предельных процессов идеального газа - Лекция, раздел Право, Газовые законы. Основные газовые процессы Наименование Процессов Характеристика ...
| Наименование процессов | Характеристика | Соотношение параметров | DU | Q | W | Выражение для первого закона термодинамики |
| Изохорный | V=const | Р/T=const | nCv (T2-T1) | nCv(T2-T1) | 0 | Q=DU |
| Изобарный | Р=const | V/T=const | nCv (T2- T1) | nCp(T2-T1) | nR(T2-T1) | Q=DU+W |
| Изотермический | T=const | PV=const | 0 | nRTln(P1/P2) nRTln(V2/V1) | nRTln(P1/P2) nRTln(V2/V1) | Q=W |
| Адиабатический | Q = 0 | PgV=const TVg-1=const TgP1-g =const | nCv(T2-T1) -[( 1/g-1)´ ´(P2V2-P2 V2)] | 0 | -nCv(T2-T1) -[(1/g-1)´ ´(P2V2-P2V2)] | W=-DU |
Лекция №9: Химическая кинетика и химическое равновесие.
Химическая кинетика – это раздел химии, изучающий скорости химических реакций. Химические реакции могут протекать с различными скоростями (от малых долей секунды до десятилетий и более продолжительных временных интервалов). При рассмотрении вопроса о скорости реакций необходимо различать гомогенные и гетерогенные реакции. Гомогенные системы состоят из одной фазы (например, любая газовая смесь), а гетерогенные – из нескольких фаз (например, вода со льдом). Фазой является часть системы, отделённая от других её частей поверхностью раздела, при переходе через которую происходит скачкообразное изменение свойств.
Скорость гомогенной реакции – это количество вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объёма системы. Скоростью гетерогенной реакцииявляется количество вещества, вступающего в реакцию или образующегося при реакции за единицу времени на единице поверхности фазы (или массы, объёма твердой фазы, когда затруднительно определение велечины поверхности твёрдого тела):
vгомог=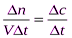 ; vгетерог=
; vгетерог=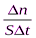 . Т.е. скорость гомогенной реакции можно определить как изменение концентрации какого-либо из веществ, вступающих в реакцию или образующихся при реакции, протекающее за единицу времени.
. Т.е. скорость гомогенной реакции можно определить как изменение концентрации какого-либо из веществ, вступающих в реакцию или образующихся при реакции, протекающее за единицу времени.
Большинство химических реакций являются обратимыми, то есть могут протекать как в прямом, так и в обратном направлениях. Рассмотрим обратимую реакцию:
aA+bB=cC+dD
Скорости прямой и обратной реакций связаны с концентрациями реагентов следующими уравнениями:
vх.р, пр=kпр[A]a×[B]b и vх.р. обр=kобр[C]c×[D]d
С течением времени скорость прямой реакции будет уменьшаться вследствие расхода реагентов Аи В и понижения их концентраций. Напротив, скорость обратной реакции по мере накопления продуктов Си D будет возрастать. Поэтому через некоторый промежуток времени скорости прямой и обратной реакций сравняются друг с другом. Установится состояние системы, в котором отсутствуют потоки вещества и энергии, называемое химическим равновесием. Все обратимые процессы протекают не полностью, а лишь до состояния равновесия, в котором из условия vх.р. пр = vх.р. обрследует:
| kпр/kобр=[C]c×[D]d/ [A]a×[B]b=K |
где K - константа химического равновесия, которая зависит от температуры и природы реагентов, но не зависит от концентрации последних. Это математическое выражение закона действующих масс, который позволяет рассчитывать состав равновесной реакционной смеси.
Важнейшими факторами, влияющими на скорость реакции, являются:
1. Природа реагирующих веществ;
2. Концентрации реагирующих веществ;
3. Температурный фактор;
4. Наличие катализаторов.
В некоторых случаях скорость гетерогенных реакций зависит также от интенсивности движения жидкости или газа вблизи поверхности, на которой реализуется реакция.
1) Влияние концентрации реагирующих веществ. Представим уравнение химической реакции в общем виде: аА+bB+…=, тогда vх.р.=k[A]a[B]b – это, по сути, математическая запись закона действующих масс, открытого опытным путём К. Гульдбергом и П. Вааге в 1864-1867 гг. Согласно указанному закону, при неизменной температуре vх.р пропорциональна произведению концентраций реагирующих веществ, причём каждая концентрация входит в произведение в степени, равной коэффициенту, стоящему перед формулой данного вещества в уравнении реакции. Величина константы скорости реакции (k) зависит от природы реагирующих веществ, температуры и наличия катализаторов, но не зависит от концентрации веществ.
2) Зависимость vх.р. от температуры и от природы реагирующих веществ.Энергией активации Еа (в кДж/моль) называют избыточную энергию, которой должны обладать молекулы для того, чтобы их столкновение могло привести к образованию нового вещества. Еа различных реакций различна. Посредством этого фактора сказывается влияние природы реагирующих веществ на vх.р.. Если Еа<40 кДж/моль (т.е. мала), то скорость такой реакции велика (например, ионные реакции в растворах, протекающие практически мгновенно). Если Еа>120 кДж/моль (т.е. очень значительна), то скорость такой реакции незначительна (например, реакция синтеза аммиака N2+3H2=2NH3 – скорость этой реакции при обычных Т вследствии высоких значений Еа настолько мала, что заметить её протекание практически невозможно).
В 1889 г. знаменитый шведский химик Аррениус вывел из опытных данных уравнение, связывающее константу скорости с температурой и энергией активации. Позднее это уравнение получило теоретическое обоснование. Согласно Аррениусу, константа скорости находится в экспоненциальной зависимости от температуры: k=kmax×exp(-Ea/RT), где R - универсальная газовая постоянная, равная 8,31 Дж/моль×К; kmax - предэкспоненциальный фактор, имеющий смысл максимально возможного значения константы скорости при нулевой энергии активации или бесконечно высокой температуре, когда все столкновения молекул реагентов становятся активными. Уравнение Аррениуса используют чаще в логарифмической форме: lnk=lnkmax-Ea/RT.
Возрастание vх.р. с ростом температуры обычно характеризуют температурным коэффициентом скорости реакции – величиной, показывающей, во сколько раз возрастает скорость рассматриваемой реакции при повышении температуры системы на 10 градусов. Температурный коэффициент (g) для разных реакций различен. При обычных температурах его значение для большинства реакций лежит в пределах от 2 до 4 (т.е. gх.р.=2-4 раза).
Катализаторами являются вещества, не расходующиеся в реакции, но оказывающие влияние на её скорость. Явление изменения скорости реакции под действием катализаторов называется катализом, а сами эти реакции являются каталитическими. Действие катализатора обусловлено снижением активационного предела химического взаимодействия, т.е. снижением величины энергии активации. Под воздействием катализаторов реакции могут ускоряться в миллионы и более раз. Более того, некоторые реакции без катализаторов вообще не реализуются. Катализаторы широко используются в промышленности.
Различают гомогенный и гетерогенный катализ. При гомогенном катализе катализатор и реагенты образуют одну фазу (газ или раствор), а при гетерогенном катализе – катализатор находится в системе в виде самостоятельной фазы. Примером гомогенного катализа служит разложение перекиси водорода на воду и кислород в присутствии катализаторов Cr2O72-, WO42- и др. Примером гетерогенного катализа является окисление диоксида серы в триоксид при контактном способе получения серной кислоты из отходящих газов металлургических производств: SO2+0,5O2+H2O=(kt)=H2SO4.
Химическое равновесие. Принцип Ле Шателье
Если система находится в равновесии, то она будет находиться в нём до тех пор, пока внешние условия сохранятся постоянными. На практике зачастую бывает важно добиться максимально возможного смещения равновесия в сторону прямой реакции (или обратной, если требуется подавить образование вредных веществ). Условия для этого выбирают на основе принципа, сформулированного известным французским учёным. Этот принцип, названный в честь французского химика Анри Луи Ле Шателье, можно сформулировать следующим образом: если на систему, находящуюся в равновесии, производится какое либо внешнее воздействие (изменяется концентрация, температура, давление), то равновесие смещается в том направлении, которое способствует ослаблению этого воздействия.
Влияние концентрации. Если увеличить концентрацию исходных веществ, то система будет стремиться быстрее их израсходовать, то есть сместится в сторону образования продуктов. И, наоборот, если в системе увеличить концентрацию продуктов, то система сместится в сторону исходных веществ.
Влияние давления. Изменение давления наиболее существенно в случае реакций, протекающих с изменением числа моль газообразных веществ.
При увеличении общего давления равновесие смещается таким образом, что общее давление снижается, то есть, смещается в направлении той реакции, которая протекает с уменьшением числа моль газообразных веществ.
Рассмотрим применение принципа Ле Шателье на примере реакции образования аммиака.
N2(gas) + ЗН2(gas) = 2NН3(gas)
Если: а) уменьшить концентрации исходных веществ N2 и Н2 б) увеличить давление равновесной смеси (сжать), то:
а) Уменьшение концентрации исходных веществ N2 и Н2 приведет к смещению равновесия справа налево, в результате концентрации N2 и Н2 вновь увеличатся за счет разложения аммиака.
б) Увеличение давления системы приведет к смещению равновесия слева направо, то есть в направлении реакции синтеза аммиака, при этом число моль газообразных веществ уменьшится (из 4-х моль исходных веществ образуется 2 моль продуктов), а соответственно уменьшится и общее давление системы.
Повышение температуры будет способствовать протеканию эндотермической реакции, идущей с поглощением тепла; понижение температуры будет способствовать протеканию экзотермической реакции, идущей с выделением тепла (DH < 0). Например, уменьшение температуры сместит равновесие реакции N2+О2=2NO (ΔН0=-180 кДж/моль) справа налево, то есть в направлении экзотермической реакции, идущей с выделением тепла. Температура системы в результате повысится.
Влияние катализатора. Катализаторы одинаково ускоряют как прямую, так и обратную реакцию, и поэтому не смещают химическое равновесие. Они способствуют более быстрому достижению равновесного состояния.
Лекция №10: Теория электролитической диссоциации. Электролиты.
Электролитами являются вещества, которые способны проводить электрический ток в растворах или расплавах. Электролиты можно классифицировать как проводники II рода. Они представляют собой вещества, распадающиеся в растворах или расплавах на ионы. Электролитами могут быть соли, кислоты и основания. Сам процесс диссоциации молекул слабых электролитов на ионы является обратимым.
Предположение Сванте Аррениуса о том, что причиной крайне высокого осмотического давления растворов электролитов является диссоциация этих электролитов на ионы, в дальнейшем было положено в основу теории электролитической диссоциации. В соответствии с этой теорией, растворяясь в воде, электролиты распадаются или диссоциируют на «+»-но и «-»-но заряженные ионы (катионы и анионы). Примеры: катионы – ионы водорода и металлов; анионы – ионы кислотных остатков и гидроксогруппы. Процесс электролитической диссоциации можно показать при помощи химических уравнений: НCl=H++Cl-. Отклонение от законов Вант-Гоффа и Рауля объяснимо распадом электролитов на ионы.
Однако теория Аррениуса не учитывала всей сложности явлений в растворах. Ей противостояла химическая, или гидратная теория растворов Д.И. Менделеева, которая базировалась на представлении о взаимодействии растворенного вещества с растворителем. Преодолеть это, на первый взгляд, противоречие двух теорий позволило предположение о гидратации ионов, впервые сделанное И.А. Каблуковым в работе «Современные теории растворов в связи с учением о химическом равновесии». Это позволило в дальнейшем объединить две указанные теории в единую.
Пусть концентрация электролита, распадающегося на 2 иона, равна С, а степень его диссоциации в данном растворе составляет a, тогда уравнение для константы диссоциации примет вид:
Кдис=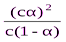 , где сa - концентрация каждого из ионов, а с(1-a) – концентрация недиссоциированных молекул.
, где сa - концентрация каждого из ионов, а с(1-a) – концентрация недиссоциированных молекул.
Это уравнение представляет собой закон разбавления Оствальда. Оно позволяет определять степень диссоциации при разных концентрациях электролита, если определена его константа диссоциации; также константу диссоциации электролита, если известна его степень диссоциации при какой-либо концентрации.
Для растворов, в которых диссоциация электролита очень мала, уравнение закона Оствальда можно упростить. В данном случае a<<1, и, следовательно, этой величиной можно пренебречь в знаменателе правой части уравнения. Тогда это уравнение примет следующий вид:
Кдис@a2·с или a=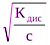 .
.
Таким образом, степень диссоциации возрастает при разбавлении раствора.
Величина Кдис электролита зависит от природы электролита и растворителя, температуры, но не зависит от концентрации раствора. Она характеризует способность электролита распадаться на ионы. Чем меньше Кдис электролита, тем слабее электролит. Значения Кдис различных электролитов приводятся в справочниках при Т=298К.
| Электролит | Константа диссоциации Кдис (при 25°С) |
| HNO2 | 4·10-4 |
| H2O2 | К1=10-12 к2=10-25 |
| H2SiO3 | К1=10-10 к2=10-22 |
| H2SO3 | К1=2·10-2 к2=10-14 |
| H2S | К1=6·10-8 к2=10-14 |
| CH3COOH | 1,74·10-5 |
| HCOOH | 1,8·10-4 |
| H2CO3 | К1=4,5·10-7 к2=4,7·10-11 |
| NH4OH | 1,8·10-5 |
| HF | 7·10-4 |
Самым слабым электролитом из приведенных является H2O2, а самым сильным – НСООН.
Вант-Гофф установил, что изотонический коэффициент i выражается дробными числами, которые с разбавлением раствора возрастают, приближаясь к целым числам. Последнее свидетельствует о неполноте диссоциации электролитов на ионы, т.к. в противном случае осмотическое давление (и пропорциональные ему величины) всегда было бы в целое число раз больше значений, наблюдаемых в растворах неэлектролитов. Аррениус объяснил это тем, что только часть электролита диссоциирует на ионы и ввел понятие степени диссоциации a (отношение числа молекул nдис, распавшихся на ионы, к общему числу молекул nS в растворе):
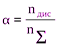 .
.
Позже было установлено, что все электролиты можно разделить на две группы: слабые и сильные. Сильные электролиты в водных растворах диссоциированны практически полностью. Понятие степени диссоциации к ним практически не применимо, а отклонение изотонического коэффициента от целочисленных значений объяснимо несколько иными причинами. Слабые электролитыдиссоциируют в водных растворах лишь частично, и в растворе имеет место динамическое равновесие между недиссоциированными молекулами и ионами.
Для оценки состояния ионов в растворе вводят понятие активности – эффективной, условной концентрации иона, в соответствии с которой он действует при химических реакциях. Активность иона а равна его концентрации с, домноженной на коэффициент активности f: a=f·с. Коэффициенты активности различных ионов различны. Они изменяются при изменении условий, например, при изменении концентрации раствора.
Если f<1, то есть взаимодействие между ионами, приводящее к их взаимному связыванию.
Если f»1, то взаимодействие между ионами носит слабый характер.
В разбавленных растворах природа ионов мало влияет на значения коэффициентов активности. Приближенно считают, что коэффициент активности данного иона зависит только от его заряда и отионной силы раствора I, которая является полусуммой произведений концентраций всех находящихся в растворе ионов на квадрат их заряда:
I=0,5·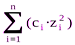 .
.
– Конец работы –
Эта тема принадлежит разделу:
Газовые законы. Основные газовые процессы
Лекция Основные классы неорганических соединений номенклатура... Основными классами неорганических соединений являются оксиды кислоты соли и... Оксиды представляют собой соединения элементов с кислородом Оксиды подразделяют на солеобразующие и несолеобразующие...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Расчет энергетических величин и выражения первого закона термодинамики для предельных процессов идеального газа
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.022 сек.
Новости и инфо для студентов