рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Медицина
- /
- Концепция А. Геррода (метаболический блок)
Реферат Курсовая Конспект
Концепция А. Геррода (метаболический блок)
Концепция А. Геррода (метаболический блок) - раздел Медицина, МЕДИЦИНСКАЯ ГЕНЕТИКА ...
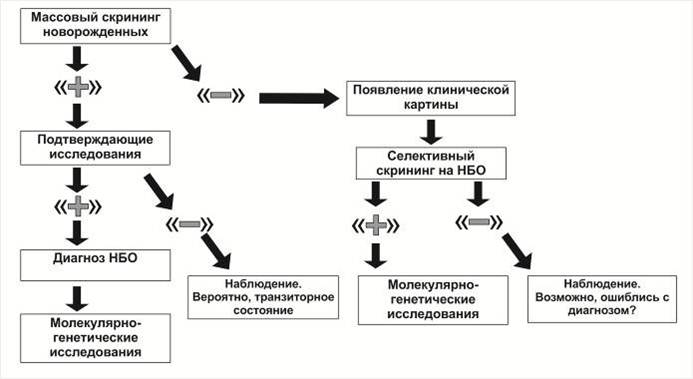 2. Схема этапной диагностики НБО
2. Схема этапной диагностики НБО
Основные описания:
Этиология НБО. Большинство синдромов, имеющих в патогенезе нарушение обмена веществ, являются моногенными. Ген кодирует фермент или белковую его часть. Распространённость таких болезней в популяции варьирует от 1 на несколько тысяч до 1 на миллионы. Другие НБО (сахарный диабет, атеросклероз и др.) имеют мультифакторную природу и встречаются значительно чаще.
Первичный уровень патогенеза НБО. Концепция Арчибальда Геррода сохраняет актуальность по настоящее время. Она описывает метаболический блок, связанный с нарушением количества или активности фермента. В результате этого в организме имеется недостаток продукта и накопление промежуточного (или побочного) метаболита, имеющего в этом случае токсический эффект.
Вторичный уровень патогенеза НБО. Патогенез любого связанного с нарушением обмена веществ синдрома должен рассматриваться на клеточном (субклеточном) уровне. По данному эффекту различают патологию лизосом (синдромы лизосомного накопления), патологию митохондрий (митохондриальные болезни), а также поражение пероксисом, комплекса Гольджи, эндоплазматического ретикулума и других органелл, фибриллярных структур клетки.
Третичный уровень патогенеза НБО. Касается изменений на уровне тканей, органов, систем, а также биохимических показателей биологических жидкостей.
Наиболее часто встречающиеся симптомы НБО. 1) Неврологические проявления – прогрессирующее отставание ребенка в психомоторном и речевом развитии, повторяющиеся приступы судорог, неустойчивая походка и двигательные нарушения, повторные бессознательные (коматозные) состояния. 2) Специфический запах и/или изменение цвета мочи, пота, слюны, кала – «мышиный» запах, запах «потных ног», «кошачьей мочи», плесени, аммиака, капусты и др. 3) Помутнение хрусталика, роговицы, специфические изменения на глазном дне. 4) Гепатоспленомегалия, расстройство стула и аппетита. 5) Изменения кожи, волос, ногтей. 6) И многое другое.
Фенилкетонури́я (ФКУ) или фенилпировиноградная олигофрения – группа ферментопатий, связанная с нарушением метаболизма и накоплением аминокислоты фенилаланин и его токсических продуктов. Фенилаланин-4-гидроксилаза катализирует превращение фенилаланина в тирозин. Резкое снижение (или отсутствие) её активности приводит к классической форме заболевания. Атипичные формы связаны с нарушением синтеза кофактора фермента – тетрагидробиоптерина. Наследуется по аутосомно-рецессивному типу. Частота встречаемости 1: 8 000.
Накопление фенилаланина (в сыворотке повышается в 10 раз) → активация вспомогательных путей распада фенилаланина → накопление в тканях токсических продуктов его обмена (фенилпировиноградной, фенилмолочной, фенилэтиламин и ортофенилацетат др. кетоновых кислот) → ↑ экскреции кетокислот с мочой → вторичные нарушения обмена тирозина и триптофана) → ↓ образование нейромедиаторов головного мозга (серотонина, ДОФА и др.) → запуск патогенетического механизма прогрессирующего слабоумия и нарушения аминокислотного равновесия организма в жидкостных средах, клетках.
Заболевание дебютирует с первых дней жизни диспепсическим синдромом. Однако, поскольку этот признак характерен для целого ряда как наследственных, так и не наследственных заболеваний, диагноз ФКУ может быть поставлен только после лабораторного подтверждения При переводе ребенка на питание без фенилаланина, диспепсический синдром проходит. При атипичной форме ответ на диетотерапию не полный. Формы определяются в процессе лечения. Умственая отсталость (олигофрения, идиотия или имбецильность, глубокая психическая инвалидность), без лечения IQ за каждые 10 недель ↓ на 5 пунктов, повышенная психомоторная возбудимость, специфическая походка, осанка, «поза портного», стереотипные движения, ↑ сухожильных рефлексов, судороги, раннее закрытие большого родничка, микроцефалия, изменения кожи, гипопигментация кожи и волос, сухость, экзема, дерматит, склеродермия. Также обнаруживаются рвота у новорождённых, светлые радужки, катаракта, «мышиный», «заплесневелый» запах тела и мочи. Своевременное начало лечения позволяет избежать развития клинических проявлений классической ФКУ. Лечение назначают до полового созревания, далее пациент придерживается сбалансированного диет-питания.
Галактоземия– нарушение обмена углеводов, при отсутствии или снижении активности фермента, превращающего галактозу в глюкозу с накоплением галактозы и продуктов ее обмена в клетках. Варианты ферментопатий: галактозо-1-фостфат-уридил трансферазы (ГАЛТ) (превращает галактозу в глюкозу), галактокиназа (УДФ-галактоза), УДФ-галактозо-4-эпимераза (отвечает за восстановление УДФ-глюкозы из УДФ-галактозы). Тип наследования аутосомно-рецессивный. Распостраненность 1:35 000 — 50 000.
Патогенез: галактоза поступает с пищей (в составе молочного сахара –лактозы) → фосфорилирование → образование галактозо-1-фосфат→наследственный дефект фермента галактозо-1-фосфат-уридилтрансферазы → превращение не происходит в связи →галактоза и галактозо-1 -фосфат накапливаются в крови и тканях → токсическое действие на ЦНС, печень, хрусталики глаз → клинические проявления.
Среди симптомов понос, рвота, отказ от пищи, непереносимость голода, снижение мышечного тонуса, поражение почек, асцит, повышение внутричерепного давления, и увеличение риска сепсиса, цирроз, катаракта у новорожденных, ЗПМР, протеинурия, гипераминоацидурия, желтуха, плохая прибавка веса, гепатоспленомегалия, расстройство функции печени, дефекты почечных канальцев, повышенная восприимчивость к инфекциям и альбуминурия в первые недели жизни. При поздней диагностике тяжелое течение.
Адреногенитальный синдром (АГС) или врожденная гиперплазия коры надпочечников (ВГКН) - наследственные нарушения, связанные с недостаточной выработкой ферментов корой надпочечников, сопровождающиеся избытком половых гормонов и недостатком глюкокортикоидов.
Чаще всего встречается дефицит 21-гидроксилазы, дефицит 11-бета-гидроксилазы, реже страдают 17-альфа-гидроксилаза, StAR (стероидогенный острый регуляторный белок), 3-бета-гидроксистероиддегидрогеназа тип II (АГС с потери соли) и др. Аутосомно-рецессивный тип наследования. Распостраненность варьирует.
Патогенез: недостаток фермента синтеза кортизола → дефицит кортизола в крови → ослабление его тормозящего влияния на продукцию кортикотропина → ↑ кортикотропина в крови → усиливается стимуляция коры надпочечников → гиперплазия сетчатой зоны → избыточная выработка андрогенов → вирилизацию детского организма. Значительный дефицит 21-гидроксилазы → ↓ продукция альдостерона → развиваться сольтеряющий синдром.
Простая вирильная форма - сочетание симптомов вирилизации у девочек (урогенитальный синус, гипертрофия клитора) и преждевременного полового созревания у мальчиков. Сольтеряющая форма — тоже плюс кризовое течение по типу острой надпочечниковой недостаточности с первых недель жизни. Гипертоническая форма — тоже плюс артериальная гипертензия. Неклассическая форма - возможны гирсутизм, акне, аменорея, бесплодие у женщин. У мужчин практически не выявляется.
Врожденный гипотиреоз (ВГ)- гетерогенная по этиологии группа заболеваний ЩЖ. Причиной могут быть наследственные дефекты биосинтеза тиреоидных гормонов (Т3, Т4), нарушение процесса преобразования прогормона в гормон или дефект белковой части гормона – тиреоглобулина, либо повреждением ЩЖ, гипоталамо-гипофизарной системы или их морфо-функциональной незрелостью во внутриутробном периоде. Распространенность 1:4000- 1:5000, у девочек в 2-2,5 раза чаще, чем у мальчиков.
Патогенез: этиологические факторы → нарушение процессов биосинтеза Т3, Т4 → усиление стимуляции работы щитовидной железы ТТГ гипофиза → развитие гипертрофии и гиперплазии щитовидной железы → не способна компенсировать тиреоидную недостаточность, ↓ или отсутствие выработки Т3, Т4 внутриутробно → микроцефалия, нарушение функции головного мозга.
Отсутствие активности ТПО → снижение захвата йодида тиреоцитами, нарушение процессов органификации йодидов → ↓ синтеза Т3, Т4. Новорожденные с ферментативными дефектами ТПО имеют при рождении очень высокий уровень ТТГ и очень низкий уровень Т4, в дальнейшем у них формируется зоб.
Среди симптомов: переношенная беременность, МТ при рождении ›4000 гр, отечное лицо, губы, веки, полуоткрытый рот с широким, "распластанный" язык, локализованные отеки в виде плотных "полушечек" в надключичиных ямках, тыльных поверхностях кистей, стоп, признаки незрелости при доношенной по сроку беременности, низкий, грубый голос при плаче, крике, позднее отхождение мекония, позднее отхождение пупочного канатика, плохая эпителизация пупочной ранки, затянувшаяся желтуха.3-4 месяце жизни, при отсутствии лечения: ЗПР, ↓ аппетит, затруднения при глотании, плохая прибавка в МТ, метеоризм, запоры, сухость, бледность, шелушение кожных покровов, гипотермия кистей и стоп, ломкие, сухие, тусклые волосы, мышечная гипотония.
Ступенчатый характер лабораторной диагностики НБО. Многие НБО можно диагностировать еще на доклиническом этапе. Существуют программы массового неонатального биохимического скрининга. Скринингу подлежат болезни, доступные для простой и достоверной биохимической диагностики, с разработанными схемами лечения, меняющими прогноз течения заболевания и имеющими отчётливую экономическую эффективность. В России согласно приказу Минздравсоцразвития России проводится обязательная диагностика пяти моногенных заболеваний. Ряд регионов России, включая Санкт-Петербург, проводят дополнительно исследования еще на 11 НБО. Сухие пятна крови на специальном бланке отправляются в учреждения, участвующие в программе. В результате формируются группы риска. Диагноз устанавливается только после подтверждающей диагностики биохимическими или молекулярно-генетическими методами.
Программы селективного скрининга существуют для отдельных групп НБО, когда на них указывают результаты клинического обследования и простых биохимических тестов, доступных для назначения врачам не генетического профиля. В медицинской практике реализуются программы обследования на аминоацидопатии и на ацидурии, другие реже. Положительный результат селективного скрининга является показанием для топических биохимических и молекулярно-генетических исследований.
Шесть принципов патогенетического лечения НБО. Основаны на концепции А. Гэррода, имеют целью компенсацию последствий «метаболического блока».
Самое распространённое лечение – ограничение субстрата в пище или увеличение недостающего продукта. При помощи коферментов можно индуцировать активность ферментов. Токсические продукты распада можно вывести либо путём их связывания, либо запустив альтернативный путь метаболизма. Ферментзаместительная терапия применяется редко, т.к. сложно доставить белок в клетки без его расщепления и реакции иммунной системы. Реализовано для болезни Гоше, мукополисахаридоза и ряда других заболеваний.
Этиотропное и симптоматическое лечение НБО. Этиотропным лечением расстройства метаболизма следует считать исключительно генотерапию. Главными проблемами которой являются доставка рекомбинантных генетических конструкций в геном пациента, высокий риск онкологических осложнений и недолговечность результата, поэтому генотерапия, как и трансплантация гемопоэтических стволовых клеток, на сегодняшний день имеет скорее экспериментальный характер. Термин «симптоматическое лечение», применительно к НБО, трактуется разными авторами неоднозначно. В наиболее широком смысле – это весь комплекс хирургических, медикаментозных, реанимационных и реабилитационных мероприятий, направленных на восстановление структуры и функции органов, предупреждение развития недостаточности определённых систем организма.
Вопросы для самоконтроля:
1) Что такое метаболизм? Какие его направления Вы знаете?
2) Что такое наследственные болезни обмена веществ?
3) Какие существуют расстройства обмена аминокислот?
4) Какие существуют расстройства обмена углеводов?
5) Какие существуют расстройства обмена липидов?
6) Какие существуют расстройства обмена азотистых оснований?
7) Какие существуют расстройства обмена стероидных гормонов, металлов?
8) Что такое «метаболический блок» по А. Герроду? Чем он опасен?
9) Какие субклеточные структуры страдают при НБО?
10) Как заподозрить патологию обмена веществ на клиническом уровне?
11) Дайте биохимическую и клиническую характеристику фенилкетонурии.
12) Дайте биохимическую и клиническую характеристику врождённого гипотиреоза.
13) Дайте биохимическую и клиническую характеристику галактоземии.
14) Дайте биохимическую и клиническую характеристику адреногенитального синдрома.
15) Перечислите подходы к патогенетическому лечению НБО, начиная с самых распространённых.
16) Сравните распространённость и перспективность этиотропного и симптоматического лечения НБО.
– Конец работы –
Эта тема принадлежит разделу:
МЕДИЦИНСКАЯ ГЕНЕТИКА
СЕВЕРО ЗАПАДНЫЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ ИМЕНИ И И МЕЧНИКОВА... Т В Харченко А Ю Петруничев Е С Шабанова... МЕДИЦИНСКАЯ ГЕНЕТИКА ПОСОБИЕ ДЛЯ ПРАКТИЧЕСКИХ ЗАНЯТИЙ СТУДЕНТОВ...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Концепция А. Геррода (метаболический блок)
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.02 сек.
Новости и инфо для студентов