рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Медицина
- /
- Методи медичної генетики
Реферат Курсовая Конспект
Методи медичної генетики
Методи медичної генетики - раздел Медицина, МЕДИЧНА ГЕНЕТИКА Тема: Клініко-Генеалогічний Метод. Цитогенетичні І Молекулярно-Генет...
Тема: Клініко-генеалогічний метод.
Цитогенетичні і молекулярно-генетичні методи.
Біохімічні методи.
Основні питання:
1. Роль параклінічних методів дослідження 
у діагностиці природженої та спадкової патології.
2. Цитогенетичний та молекулярно-цитогенетичні методи. Показання до проведення цитогенетичних досліджень.
3. Клініко-генеалогічний метод.
4. Методика складання родоводу.
5. Типи успадкування.
6. Мітохондріальна спадковість.
7. Біохімічні методи. Показання до проведення досліджень.
8. Молекулярно-генетичні методи. Показання та можливості методу.
Чим глибше аналізується природа спадковості людини, тим більш це реалізується в методах діагностики, лікування і профілактики хвороб. При вивченні спадкових ознак людина виступає як складний об'єкт генетичних досліджень.
Виникають певні труднощі в аналізі спадковості і мінливості, які обумовлені:
• неможливістю застосування направлених схрещувань (гібридологічного методу) для генетичного аналізу;
• неможливістю експериментального отримання мутацій;
• пізнє настання статевої зрілості;
• малою чисельністю нащадків;
• неможливістю забезпечення однакових контрольованих умов для розвитку нащадків від різних шлюбів;
• недостатньо точною реєстрацією спадкових ознак;
• порівняльне великим числом (2п = 46) хромосом.
Сучасна клінічна медицина вже не може обійтися без генетичних методів. Для вивчення спадкових ознак у людини використовують різні біохімічні, морфологічні, імунологічні, електрофізіологічні методи. Лабораторно-генетичні методи діагностики завдяки прогресу генетичних технологій можуть бути виконані на малій кількості матеріалу, який можна пересилати по пошті (декілька випущених крапель крові на фільтрувальному папері, або навіть на одній клітині, взятій на ранній стадії розвитку.
У вирішенні генетичних завдань використовують такі методи:
- цитогенетичний,
- генеалогічний,
- близнюковий,
- популяційно-статистичний,
- біохімічні,
- молекуляро-генетичні.
Клініко-генеалогічнийметод є найбільш універсальним методом у медичній генетиці. Він широко застосовується при рішенні наступних теоретичних і прикладних проблем:
1) для встановлення спадкового характеру ознаки;
2) при визначенні типу успадкування і пенетрантності гена;
3) при аналізі зчеплення генів і картування хромосом;
4) при вивченні інтенсивності мутаційного процесу;
5) при вивченні механізмів взаємодії генів;
6) при медико-генетичному консультуванні.
Суть методу зводиться до виявлення родовідних зв'язків і простежуванню ознаки або хвороби серед близьких і далеких родичів. Технічно він складається із двох етапів:
§ складання родоводу,
§ генеалогічного аналізу.
Перше завдання при аналізі родоводу - встановлення спадкового характеру ознаки. Після того як буде виявлений спадковий характер ознаки (хвороби), необхідно встановити тип успадкування. Для цього використовуються принципи генетичного аналізу й різні статистичні методи обробки даних не з однієї, а з багатьох родоводів. Неважко зрозуміти, що в більшості випадків прості розрахунки співвідношення хворих дітей до числа здорових дадуть неправильне уявлення про тип успадкування, тому, що, наприклад, при рецесивному захворюванні в поле зору лікаря не потраплять родини носії, у яких народилися тільки здорові діти. Практично реєстрація починається від хворого нащадка. У розрахунки співвідношення числа хворих і здорових дітей потрібно вводити виправлення на частку невиявлених родин.
Збір відомостей про родину починається із пацієнта, що консультується або із пробанда. Пацієнт, що консультується - це особа, яка звернулася до лікаря або, що потрапила в поле зору дослідника. Пробанд - це хворий або носій досліджуваної ознаки. У багатьох випадках особа, що консультується, й про банд співпадають. Діти однієї батьківської пари називаються сибсами (брати та сестри). Родиною у вузькому змісті називають батьківську пару і їхніх дітей, але іноді й більше широке коло кревних родичів, хоча в останньому випадку краще використовувати термін «рід».
Символи, використовувані при складанні родоводів:
1-  особа чоловічої статі;
особа чоловічої статі;
2- особа жіночої статі
3- стать не відома;
4- шлюб;
5- родинний шлюб;
6- сибси;
7- монозиготні близнюки;
8- дизиготні близнюки;
9- викидень;
10- аборт;
11- мертвонароджений;
12- бездітний шлюб;
13- гетерозиготна
носителька мутантного
гена в X-хромосомі;
14- померлі;
15- пробанд.
Залежно від мети дослідження родовід може бути повним або обмеженИМ. Бажано, звичайно, прагнути до найбільш повного складання родоводів за усіми напрямками. Завдання це не така легке, як може здатися на перший погляд. Чим більше поколінь утягується в родовід, тим він ширше. Це спричиняє неточність одержуваних відомостей і, отже, неточність родоводу в цілому.
Приклад родоводу (позначення стандартні):
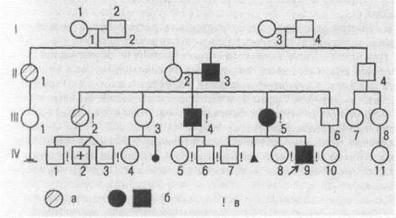
а - хворі діабетом;
б - хворі нейро-
фіброматозом;
в - особисто обстежені.
Складання родоводу супроводжують коротким записом про кожного член родоводу з точною характеристикою його споріднення стосовно пробанду (легенда родоводу). Надалі для наочності (або при публікації) родовід зображують графічно. Якщо розглянутих ознак у родоводу багато, то можна прибігати до буквених або штрихових розходжень усередині символів. Зображення родоводу обов'язково супроводжується описом позначень під малюнком.
На додаток до опису малюнка варто підкреслити наступне. Покоління позначають римськими цифрами зверху долілиць. Звичайно цифри ставлять ліворуч від родоводу. Арабськими цифрами нумерують потомство одного покоління (весь ряд) ліворуч праворуч послідовно. Брати й сестри розташовуються в родоводі в порядку народжень. Таким чином, кожний член родоводу має свій шифр, наприклад ІІ-2, ІІІ-8.
У тих випадках, коли дружина або чоловік не обстежений на наявність розглянутої ознаки і його родовід не приводиться, бажано не зображувати його взагалі. Нанесення такого значка в родовід не несе ніякої інформації, а тільки утрудняє сприйняття основної частини родоводу.
Всі індивіди повинні розташовуватися строго по поколіннях в один ряд. «Підвішування» символів між рядами поколінь є досить грубою помилкою.
Якщо родовід дуже велика, то різні покоління розташовуються не горизонтальними рядами, а концентричними.
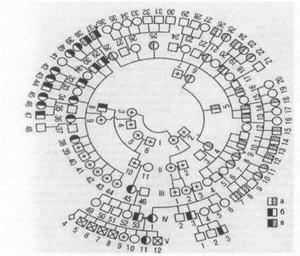 Приклад великого родоводу з концентричним розташуванням поколінь:
Приклад великого родоводу з концентричним розташуванням поколінь:
а - хворі гемоглобінозом Е;
б - хворі таласемією;
в - хворі гемоглобінозом Е та таласемією.
При застосуванні генеалогічного методу в родоводі важливо відзначати обстежених на наявність ознаки (це можна дорівняти також до одержання відомостей з об'єктивного джерела, наприклад, з історії хвороби) і необстежених, відомості, про які почерпнуті з відповідей пробанда або родичів, а також з анкет. Грубою помилкою є штучне вкорочення ланок родоводу через труднощі обстеження осіб 2-й і 3-й ступеня споріднення. Причому не завжди вказується, у кого зі членів родоводу дійсно не було родичів, а в кого просто не зібрані відомості.
Одержання відомостей про родичів - завдання непросте. По-перше, не всі пацієнти знають про хвороби родичів, по-друге, нерідко вони приховують сімейні випадки через фальшивий сором або, навпаки, «відкривають» їх у родичів чоловіка, намагаючись «звалити провину» у виникненні хвороби в ребенка на чоловіка.
Докладне клініко-генеалогічне дослідження проводиться у всіх випадках, коли при первинному клінічному огляді виникає підозра на спадкову хворобу. Варто підкреслити, що мова йде про докладне обстеження членів родини. На відміну від первинних елементів сімейного аналізу, які застосовуються при будь-якому первинному огляді хворого, при докладному клініко-генеалогічному обстеженні мова йде вже про цілеспрямоване глибоке обстеження членів родини.
Хвороби з аутосомно-домінантним типом спадкування.
Цей тип спадкування характеризується тим, що для розвитку хвороби досить успадкувати мутантний аллель від одного з батьків. Для цієї групи захворювань характерний ряд загальних ознак:
1. Захворювання зустрічається в кожному поколінні родоводу, що називають передачею хвороби по вертикалі. Іноді може бути «проскакування поколінь» через пізню маніфестацію або у зв'язку з різної пенетрантностью й експресивністю ознаки.
2. Співвідношення хворих і здорових наближається до 1:1.
3. У здорових дітей хворих батьків діти здорові.
4. Кількість хворих хлопчиків і дівчинок однаково.
5. Хворі чоловіки й жінки з однаковою частотою передають патологічний ген своїм дітям.
6. У гомозигот, породжених від двох хворих батьків, захворювання протікає важче, ніж у гетерозигот.
При важких захворюваннях, коли у хворих знижена можливість мати потомство, родоводи можуть бути нетиповими, як і у випадках, коли мутації виникають уперше в гаметах (спорадичні випадки).
Прикладами захворювань із аутосомно-домінантним спадкуванням можуть служити наступні:
§ синдром Марфана,
§ нейрофіброматоз,
§ синдром базальноклітинного невуса,
§ ахондроплазія,
§ недосконалий остеогенез,
§ хвороба Альцгеймера,
§ міотонічна дистрофія,
§ полікістоз нирок дорослого типу та ін.
Відзначається високий клінічний поліморфізм домінантно наслідуваних захворювань навіть серед членів однієї родини: так, при нейрофіброматозе в одних хворих у родині можуть бути множинні нейрофіброми, а в інших - лише одиничні шкірні прояви. Також відрізняються строки маніфестації хвороби, навіть у межах однієї родини.
Хвороби з аутосомно-рецесивным типом спадкування.
Захворювання з даним типом спадкування проявляються тільки в гомозигот. Гетерозиготи не мають клінічних проявів захворювання.
Для аутосомно-рецесивних захворювань характерно наступне:
1. Батьки звичайно клінічно здорові, однак є гетерозиготними носіями патологічного гена.
2. Ризик по захворюванню для сибсів хворого становить 25%.
3. Якщо хворі обоє батьків, то всі діти будуть хворими.
4. У шлюбі хворого зі здоровим народжуються здорові діти (якщо здоровий не є гетерозиготою), які будуть носіями патологічної ознаки.
5. У шлюбі хворого з носієм мутантного алеля народжуються 50% хворих дітей, що імітує домінантне спадкування (псевдодомінювання).
6. Обидві підлоги дивуються однаково.
7. Ніж рідше зустрічається мутантний ген у популяції, тим частіше батьки хворого ребенка є кровними родичами. При такому шлюбі ризик по захворюванням з аутосомно-рецесивным типом успадкування значно перевищує загальнопопуляційний.
Найбільше часто зустрічаються шлюби, де обоє батьки є гетерозиготами та ризик появи хворого ребенка становить 25%. Однак варто мати на увазі, що це популяційний ризик, а співвідношення хворих і здорових дітей у кожній окремо взятій родині може бути різним.
Шлюби, коли обоє батька є гомозиготами, дуже рідкі.
У родинах, де у хворих батьків (наприклад, у альбіносів) народжувалися здорові діти, така невідповідність пояснюється мутаціями в різних генах, і такі діти є подвійними гетерозиготами, або компаунд-гетерозиготами.
За аутосомно-рецесивним типом успадковуються багато вроджених порушень обміну речовин. Прикладами захворювань із цим типом спадкування можуть бути:
§ муковісцидоз,
§ фенілкетонурія,
§ галактоземія,
§ хвороба Вільсона-Коновалова,
§ адреногенітальный синдром,
§ мукополісахаридози,
§ синдром Мекеля.
Варто сказати, що для багатьох із цих захворювань розроблені методи пренатальной діагностики, що проводиться в родинах, де батьки є гетерозиготними носіями патологічної ознаки.
Хвороби з Х-зчепленим домінантним типом спадкування.
Особливості цього типу спадкування обумовлені тим, що в жінок дві Х-хромосоми, а в чоловіків одна. Тому жінка, успадкувавши від одного з батьків патологічний ген, є гетерозиготою, а чоловік гомозиготою. Основні характеристики родоводів при цьому типі спадкування наступні:
1. Вражаються й чоловік, і жінки, але хворих жінок більше.
2. Хворі жінки передають патологічний алель 50% дочок і 50% синів.
3. Хворий чоловік передає патологічний алель всім дочкам і не передає синам, оскільки останні не одержують від батька Х-хромосому.
4. У середньому захворювання у жінок протікає менш важко, тому що вони гетерозиготни.
5. Якщо хвороба важка й летальна для гомозигот, то всі хлопчики гинуть. Хворими бувають тільки дівчинки.
За цим типом успадковуються наступні захворювання:
§ фосфат-діабет (або вітамін Д - резистентний рахіт),
§ хвороба Блоха-Сульцбергера (нетримання пігменту),
§ рото-обличчя-пальцьовий синдром.
Хвороби з Х-зчепленим рецесивным типом спадкування.
При захворюваннях із цим типом спадкування жінки практично завжди є гетерозиготами, тобто фенотипично здорові, але є носіями. У випадку, якщо репродукція у хворих порушена, характерні наступні риси спадкування:
1. Хворіють тільки чоловіки.
2. Жінки є гетерозиготними носіями патологічного гена.
3. У частині випадків захворювання є результатом неомутації в Х-хромосомі матері
4. Сестри хворих братів в 50% випадків можуть бути носіями патологічного алеля. При цьому вони в 50% випадків передають ген синам, які будуть хворі, і в 50% дочкам, які також можуть будуть носіями.
5. Здорові чоловіки не передають хвороби.
Якщо репродукція при даній хворобі не порушена (гемофілія, недостатність глюкозо-6-фосфатдегидрогенази), то спадкування характеризується в такий спосіб:
1. Частка успадкованих випадків більше 23.
2. Хворі чоловіки передають патологічний алель всім своїм дочкам, і не передають синам.
3. Усі дочки хворих чоловіків є носіями патологічного гена.
4. У шлюбі жінки-носія із хворим чоловіком 50% дочок хворі, 50% синів хворі, 50% здорові.
Рідко гетерозиготні жінки можуть бути хворими у зв'язку з гетерохроматинизацією хромосоми з нормальним алелем.
Успадковуються за X-зчепленим рецесивним типом:
§ гемофілія,
§ м'язова дистрофія Дюшена,
§ синдром Хантера (мукополісахаридоз 2 типу),
§ дальтонізм,
§ синдром Леша-Ніхана,
§ ангідротична ектодермальна дисплазія.
Голандричне, або зчеплене з Y-хромосомою спадкування.
Рідкий тип спадкування, при якому ознака передається від хворого батька всім хлопчикам. Природно, що при порушенні формування семеників або сперматогенезу хворі безплідні; у цих випадках захворювання є результатом неомутації.
На Y-хромосомі локалізований ряд генів:
§ ген, детерминуючий розвиток семеників, відповідальний за сперматогенез (фактор азооспермії),
§ ген, що контролює інтенсивність росту тіла, кінцівок і зубів,
§ ген, що визначає оволосение вушних раковин.
Мітохондріальна спадковість.
Особливістю цього типу спадкування є те, що мітохондрії перебувають у цитозолі клітин. Спермії не мають мітохондрій, тому спадковий матеріал передається із цитоплазмою тільки через яйцеклітини, тобто має місце материнський тип спадкування. Кожна яйцеклітина містить 25000 мітохондрій, і кожна мітохондрія містить кільцеву ДНК. Описано мутації цілого ряду генів мітохондрій. Особливості цієї спадковості:
1. Хвороба передається тільки від матері.
2. Чоловіки й жінки вражаються з однаковою частотою.
3. Хворі батьки не передають захворювання своїм дітям.
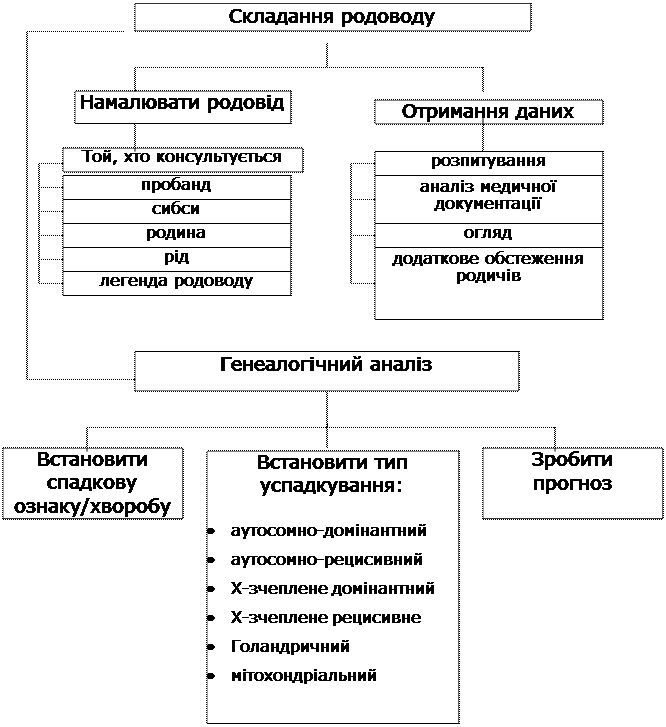
Граф логічної структури теми:
«Клініко-генеалогічний аналіз»
Цитогенетичний метод. Цитогенетичний аналіз дозволяє записувати діагноз спадкового захворювання у вигляді каріотипічної формули. Цитогенетичний метод (метод хромосомного аналізу) ґрунтується на мікроскопічному дослідженні структури і кількості хромосом. Він набув широкого застосування в 20-х роках XX ст., коли було отримано перші відомості про кількість хромосом у людини. У 30-х роках ідентифіковано перших 10 пар хромосом. У 1956 р. шведські вчені Дж.Тийо і А.Леван вперше довели, у людини 46, а не 48 хромосом.
Цитогенетичний метод використовують для:
• вивчення каріотипів організмів;
• уточнення числа хромосомних наборів, кількості і морфології хромосом для діагностики хромосомних хвороб;
• складання карт хромосом;
• для вивчення геномного і хромосомного мутаційного процес
• вивчення хромосомного поліморфізму в людських популяція
Стандартні цитогенетичні методи:
1) ФТА - культивування лімфоцитів;
2) диференційне забарвлення хромосом – Q, G, R, С.
3) NOR - забарвлення ядерце-утворюючих ділянок акроцентричних хромосом.
Хромосомний набір людини містить велику кількість хромосом, основні відомості про які можна отримати при вивченні їх в метафазі мітозу і профазі - метафазі мейозу. Клітини людини для прямого хромосомного аналізу отримують шляхом біопсії кісткового мозку і гонад, або непрямим методом - шляхом культивування клітин периферичної крові (лімфоцити), коли отримують значну кількість метафаз. Непрямим методом досліджують також клітини амніотичної рідини або фібробласти, отримані при амніоцентезі або біопсії хоріона клітини абортусів, мертвонароджених та ін.
Останнім часом всі дослідження в цитогенетиці людини проводять із застосуванням методів диференційного забарвлення хромосом; розроблено нові методики забарвлення хромосом, які дозволяють відрізнити кожну хромосомну пару (диференціальне забарвлення хромосом). Існує декілька способів забарвлення: Q, G, С, R. У вирішенні питань діагностики хромосомних хвороб різні методи диференціального забарвлення застосовують у комбінації.
Завдяки диференціальному забарвленню хромосом можна виявити незначні хромосомні поломки: невеликі делеції, транслокації та ін. Хромосомні зміни виявляють, досліджуючи каріотип дорослого організму, в клітинах амніотичної рідини і в клітинах хоріону для діагностики хромосомних захворювань плода.
Одним із останніх сучасних методів уточнюючої цитогенетичної діагностики є високорозміщуючий молекулярно-цитогенетичний метод, який отримав назву гібридизація in situ. Роздільна здатність методу складає 5х104п.о., тому він дозволяє досліджувати хромосомні сегменти довжиною від 5х10 (4) до 5х10 (6) п.о. Для хромосоми або її ділянки, що вивчається, синтезують комплементарну послідовність ДНК і приєднують до неї мітку. "Міткою” можуть бути різні речовини, зокрема радіоактивні і флуоресцентні (флуорохроми). "Помічену" послідовність ДНК називають зондом. Зонд "знаходить" комплементарну послідовність в хромосомному наборі і приєднується до неї. Потім надлишок видаляють і проводять визначення сигналу гібридизації.
Відома модифікація цього метода, - флуоресцентна гібридизація in situ (FISH). Для проведення Fish, зонди різняться не тільки специфічністю по довжині, але і за способом мічення. Метод використовують не тільки для визначення розташування гена, але і для розшифрування складних хромосомних перебудов (уточнення хромосомних фрагментів, виявлення хромосомного мозаїцизму, розташування місць розривів при транслокації та ін.).
Найбільш частіше проводять наступні цитогенетичні дослідження:
- Дослідження хромосом у лімфоцитах периферичної крові на стадіях метафази і прометафази з використанням диференціального фарбування (G-, С-, R- і Q- методи)
- Виявлення специфічного сайту ламкості X-хромосоми.
- Визначення Y-хроматину в клітинах крові
- Визначення X-хроматину в клітинах слизової оболонки порожнини рота (половий хроматин)
Біохімічні методи.Відомо понад 1000 спадкових захворювань обумовлених дефектами обміну речовин. Згідно з класифікацією ВОЗ спадкові дефекти обміну речовин поділяються на порушення:
• амінокислотного обміну;
• вуглеводного обміну;
• ліпідного обміну;
• стероїдного обміну;
• пуринового і піримідинового обмінів;
• аномалії обміну металів;
• обмін речовин в еритроцитах і порушення їх обміну та ін.
Для вивчення ферментативних порушень використовують методи ензимології. Важливе значення мають не тільки кількісні зміни активності фермента, але і якісні відмінності у функціонуванні нормального і зміненого фермента.
Застосування біохімічних досліджень показано при підозрі на всі спадкові хвороби обміну речовин (CХО) і інші форми з точно встановленим дефектом первинного генного продукту або ланки. Біохімічні методи дозволяють виявити нестачу певних сполук або надлишок їх попередників, і перш за все хроматографічні методи (хроматографія на папері, іонообмінних смолах, в тонких шарах, газорідинна хроматографія, методи електрофорезу, імуноелектрофорезу та ін.). Поєднання їх із навантажувальними пробами значно підвищує інформативність дослідження.
СХО біохімічне можуть бути діагностуванні за допомогою:
• визначення структури аномального білка (структурних білків або ферментів, таких, як аномальні гемоглобіни, несправжня холінестераза).
• визначення проміжних продуктів обміну, які з'являються внаслідок генетичного блоку прямої реакції обміну. Це найбільш поширений метод діагностики різних ензимопатій.
Найбільш відомі біохімічні методи досліджень:
1. Діагностика порушень амінокислотного обміну. Для визначення змін обміну амінокислот досліджують кров або сечу пацієнта.
2. Діагностика глюкозурій. Відомо понад 15 дефектів обміну вуглеводів. При цьому порушується або синтез ферментів вуглеводного обміну, або транспорт вуглеводів в ниркових канальцях, або всмоктування їх в кишках. Зазначені порушення діагностують біохімічними тестами:
- скринінг-тести (спадкові хвороби обміну речовин);
- напівкількісне визначення спектру вільних амінокислот у крові і сечі за допомогою тонкошарової хроматографії (порушення амінокислотного обміну);
- визначення уропорфирину, копропорфирину і порфобіліногену (спадкові порфирії);
- визначення сечової кислоти (синдром Леш-Ніхана);
- визначення активності альфа-1-антитрипсину (дефіцит альфа-1-антитрипсину);
- визначення іонів хлору в потовій рідині (муковісцидоз);
- визначення активності галактозо-1-фосфат уриділтрансферази (галактоземія).
3. Проба на мукополісахариди. Ряд спадкових захворювань характеризуються появою в сечі мукополісахаридів. Їх виявляють пробою з ортотулоїдином або тонкошаровою хроматографією. Зразки матеріалу (кров, сеча) можна наносити на диски фільтрувального паперу і пересилати в центральні біохімічні лабораторії для проведення аналізу на:
- визначення вмісту загальних глікозаміногліканів у сечі (ЦПХ-тест) (мукополісахаридози).
- фракціонування ГАГ за допомогою тонкошарової хроматографії (мукополісахаридози).
- визначення спектру олігосахаридів у сечі за допомогою тонкошарової хроматографії (олігосахаридози).
- визначення активності лізосомних ферментів у лейкоцитах і плазмі крові:
§ арилсульфатаза А (метахроматична лейкодистрофія)
§ арилсульфатаза В (синдром Марото-Ламі)
§ гексозамінідаза-А і –В (синдроми Тея-Сакса і Сандхоффа)
§ галактозідаза (синдром Фабрі)
§ галактозідаза (генералізований гангліозидоз)
§ глюкуронідаза (синдром Слая)
§ глюкозідаза (синдром Помпе)
§ глюкозідаза (синдром Гоше).
Показання до біохімічного генетичного дослідження:
1. Затримка психічного розвитку і розумова відсталість; іноді в поєднанні з патологією інших органів і систем.
2. Порушення поведінки, розгальмування або аутизм, алалія.
3. Судорожний синдром, м'язова гіпо- або гіпертонія, порушення координації, летаргічний стан, гіпертермія.
4. Зниження зору або сліпота, зниження слуху або глухота.
5. Деформації кістяка, малорухомістьсть або гіпермобільність суглобів.
6. Гіпо- або гіперпігментація, підвищена фоточутливість шкіри, жовтяниця, шкірні висипання, екзема.
7. Гепато- та спленомегалія.
8. Нестерпність окремих харчових продуктів і лікарських препаратів. Поява після їхнього прийому блювоти, діареї, зниження апетиту, жирного випорожнення, ознак гемолітичної анемії.
9. Ознаки сечокислого діатезу, у т.ч. нирково-кам'яної хвороби в дитячому віці.
10. Незвичайний колір і запах сечі, незвичайний запад тіла, дихання.
Обов'язкові лабораторні дослідження крові при підозрі на спадкові порушення обміну речовин з гострим початком:
§ Загальний аналіз крові
§ Гази крові
§ Бікарбонати
§ Електроліти
§ Сечовина
§ Глюкоза
§ Фосфор
§ Кальцій
§ Аміак
§ Аспартат і аланінамінотрансфераза
§ Лактат
§ Амінокислоти
– Конец работы –
Эта тема принадлежит разделу:
МЕДИЧНА ГЕНЕТИКА
МЕДИЧНА ГЕНЕТИКА... самостійна підготовка студентів...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Методи медичної генетики
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.033 сек.
Новости и инфо для студентов