рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Образование
- /
- Органическая реакция и синтетический метод
Реферат Курсовая Конспект
Органическая реакция и синтетический метод
Органическая реакция и синтетический метод - раздел Образование, ОРГАНИЧЕСКИЙ СИНТЕЗ Термину «Синтетический Метод» Трудно Дать Строгое Определение...
Термину «синтетический метод» трудно дать строгое определение, но не трудно описать смысл этого понятия. Идеальный синтетический метод может быть уподоблен оператору в математике, «черному ящику» в кибернетике или преобразователю в радиотехнике — короче говоря, любой логической или технической структуре, которая безотказно производит над объектом некоторое однозначно определенное преобразование. Синтетический ме-'1од — некоторая более или менее стандартизированная последовательность операций, результатом которой является строго определенное преобразование структуры исходных соединений независимо от частных особенностей строения последних. Внутри этого «черного ящика» обязательно присутству-тй*г одна или несколько реакций, определенный набор реагентов, растворителей и катализаторов, те или иные процедуры выделения и т. д. ?■ Разумеется, главный критерий ценности синтетического метода — это характер достигаемого с его помощью превращения. Это превращение должно 'быть целенаправленным и, как правило, вести от более доступных предшественников к менее доступным соединениям. Например, ароматические углеводороды — в целом доступные соединения, получаемые в больших количествах при переработке угля и нефти, и именно они используются в синтезе многих тысяч различных производных ароматического ряда. Основной реакцией во многих из этих синтезов является электрофильное ароматическое замещение (см. выше, например, ионное бромирование толуола или ацетилирование толуола по Фриделю—Крафтсу). Именно в силу этих причин реакции этого типа были исследованы подробнейшим образом и доведены до ■уровня синтетических методов почти 100%-ной надежности.
Ключевая реакция метода может предполагать использование нестабильных реагентов или интермедиатов. Тем не менее, совокупность нескольких Элементарных реакций, если они увязываются в стройную последовательность, начинающуюся с подходящих исходных веществ, уже может составить основу хорошего синтетического метода. Так, например, реакция маг-нийорганических соединений (реактивов Гриньяра) с диоксидом углерода (одна из многих реакций Гриньяра) представляет собой надежный путь синтеза карбоновых кислот. Однако реактивы Гриньяра могут быть не очень устойчивыми, почти не подлежат хранению и лишь немногие из них являются коммерчески доступными. К счастью, их совершенно не обязательно готовить заранее, а можно получать непосредственно в реакционной колбе взаимодействием магния с легкодоступными галогенопроизводными и использовать сразу же для реакции с СО2. Поэтому последовательность трех реакций, показанных ниже, служит основой превосходного метода синтеза карбоновых кислот из органических галогенидов, в результате которого углеродная цепь удлиняется на один атом:
1. RHal + Mg → R-MgHal
2. R-MgHal + CO2→ R-COOMgHal
3. R-COOMgHal + H3O+ → R-COOH
Эта последовательность имеет практически универсальную применимость, и поэтому ее вполне правомерно представлять в терминах «черных ящиков» как результат применения оператора «Метод Гриньяра» к системе RHal:
RHal → [Метод Гриньяра] → R-COOH
Каким же требованиям должна отвечать органическая реакция, чтобы служить основой хорошего синтетического метода? Прежде всего, эта реакция должна быть общей, что подразумевает, что в нее может вступать не одно какое-то соединение и даже не группа родственных соединений, а большинство или — в пределе — все возможные соединения, содержащие определенный элемент структуры — функциональную группу, способную к данной реакции. Иными словами, общность предполагает слабую зависимость хода реакции по этой функции от конкретных особенностей строения остальной части молекулы. Именно общность данной реакции позволяет с уверенностью предсказывать ее результат в применении к новым еще не изученным соединениям, а это та ситуация, которая чаше всего возникает при планировании и осуществлении многостадийных синтезов. Общность реакции позволяет нам пользоваться при ее схематическом описании символами типа R, ибо при этом делается молчаливое предположение о том, что содержание схемы не зависит от конкретной природы остатка, изображаемого таким символом.
Открытие новой реакции может явиться результатом счастливой находки или рационально направленного поиска, но так или иначе оно всегда первоначально охватывает очень ограниченное число примеров. Поэтому для оценки действительного значения этого открытия всегда требуется оценить общность и границы применимости реакции на возможно более разнообразном наборе примеров. Поясним сказанное на истории разработки такого хорошо известного метода построения циклических систем, каким является реакция Дильса—Альдера.
 Z – электроноакцепторная группа
Схема 2.7
Z – электроноакцепторная группа
Схема 2.7
|
Сущность этой реакции заключается в образовании производных циклогексена путем присоединения олефинов к сопряженным диенам (схема 2.7). Отдельные случаи такого циклоприсоединения, как, например, циклодимеризация изопрена, были обнаружены сравнительно давно, еще в начале XX в., но именно как изолированные, частные наблюдения, которые не привлекли особого внимания химиков. Подлинное открытие этой реакции состоялось лишь в 1928 г. благодаря фундаментальным исследованиям Дильса и Альдера [2а]. В этих работах самым обстоятельным образом были выяснены требования к диену и диенофилу (олефину), показана почти универсальная применимость реакции в рамках соблюдения этих требований, установлены основные закономерности ее структурной и пространственной направленности и найдены оптимальные условия проведения. Примечательным было то обстоятельство, что с самого начала своих исследований авторы осознавали огромный синтетический потенциал этой реакции, и все изучение ее особенностей по сути дела было подчинено решению задачи разработки на ее основе общего препаративного метода. В результате интенсивнейших исследований в этой области не только удалось открыть по сути дела но-.вый класс органических реакций — реакции циклоприсоединения, но и со-.здать один из самых эффективных и безотказных методов органического «синтеза, что во многом предопределило успешное решения целого ряда задач полного синтеза сложнейших соединений. Эта работа по справедливости была увенчана в 1950 г. Нобелевской премией, а сам метод, что, может быть, еще более почетно, навсегда вошел в золотой фонд органической химии под названием «реакция Дильса—Альдера».
Вот еще пример несколько иного рода. Упомянутый выше синтез карбоновых кислот является лишь одним из серии мощных синтетических методов, основанных на реакции Гриньяра, открытой в конце XIX в. Критическим для успешного проведения этой реакции, как было изначально установлено Гриньяром, является использование диэтилового эфира как растворителя на стадии приготовления магнийорганических производных (реактивов Гриньяра). Область препаративного применения реакции Гриньяра оказалась чрезвычайно широкой, поскольку галогенопроизводные почти любых типов могли быть превращены в соответствующие магниевые производные. «Почти» относилось к одному конкретному классу, а именно к винилгалогенидам, которые не удавалось превратить в реагенты Гриньяра в этих условиях, что существенно ограничивало область применения реакций всего этого класса. Решение — и необычно простое — было найдено Норманом [2Ь] в 1950 г., который показал, что превращение винилгалогенидов в соответствующие реагенты Гриньяра может ,быть осуществлено легко и эффективно, если в качестве среды для реакции использовать не диэгиловый эфир, а тетрагидрофуран. Благодаря этой, казалось бы не очень принципиальной, вариации в методике проведения реакции удалось сделать метод Гриньяра существенно более общим, распространив его применимость и на возможность переноса алкенильной группы.
Конечно, общность реакции в первую очередь определяется ее химизмом, т. е, природой происходящих превращений. Однако не так уж редки случаи, когда источником ограничений служат обстоятельства не фундаментального, а чисто технического характера. Очень нередки, к примеру, ограничения, накладываемые растворимостью компонентов реакционной системы. Типичны в этом отношении затруднения, возникающие при необходимости провести реакцию не растворимою в воде органического вещества с вешествами неорганическими (например, солями), не растворимыми в обычных органических растворителях. Так, например, упоминавшееся выше окисление перманганатом калия, как и ряд других сходных случаев окисления неорганическими окислителями, — это очень эффективные реакции, в принципе применимые для широкого круга субстратов. Однако перманганат калия практически нерастворим в большинстве органических растворителей, и поэтому в классических методиках требовалось проводить его реакции с во-донерастворимыми органическими соединениями в гетерогенной среде, т. е. в условиях, далеких от оптимальных. В 1960-х годах было найдено общее решение этой и других сходных проблем, связанных с фазовой разобщенностью реагентов, о сути которого мы сейчас расскажем.
Зададимся вопросом, а почему, собственно говоря, перманганат калия растворим в воде, но совершенно нерастворим, например, в бензоле? Причины этого различия хорошо известны: при растворении в воде энергетические затраты на разрушение кристаллической структуры КМnО4 с лихвой перекрываются благодаря энергии сольватации ионов К+ и МnО4- полярными молекулами воды, но ничего подобного не может происходить в среде такого малополярного растворителя, как бензол, поскольку молекулы последнего неспособны эффективно сольватировать ионы. Это противоречие удалось преодолеть, используя третий компонент — вешество, растворимое в бензоле и в то же время способное эффективно выполнять роль сольватной «шубы» для ионов.
 Схема 2.8
Схема 2.8
|
Здесь речь идет о циклических полиэфирах 17 — краун-эфирах, открытых Педерсеном [2с] в 1960-х годах (см. разд. 4.2.2). Полость внутренней части молекулы 17 достаточна по размеру для того, чтобы там разместился ион калия, а наличие шести атомов кислорода обеспечивает возможность образования прочной системы координационных связей, как это показано в структуре 18, вполне заменяющих гидратную оболочку (схема 2.8). Поэтому комплексы типа 18 уже достаточно хорошо растворимы в органических растворителях, в чем легко убедиться с помощью простого эксперимента: если взять двухфазную систему ярко-окрашенный водный раствор перманганата калия — бесцветный бензол и добавить в нее небольшое количество краун-эфира 17, то бензольный слой немедленно окрашивается в интенсивный малиновый цвет. Понятно, что в такой системе окисление органического вещества протекает несравненно эффективнее и неудивительно, что использование этого, казалось бы чисто технического, приема коренным образом изменило возможности синтетического применения окисления неорганическими солями в органической химии.
Тот же прием, называемый межфазным катализом (или катализом фазового переноса), также эффективно используется для форсирования множества других реакций, требующих участия неорганических реагентов [2d]. Помимо упомянутых краун-эфиров типа 17, существует еще ряд других типов соединений, также способных служить активными катализаторами фазового переноса (подробнее об этом см. разд. 4.2.2.).
Однако общность реакции — это, хотя и необходимая, но далеко не достаточная характеристика для оценки значимости реакции как синтетического метода. Классическая органическая химия, в том виде как она сложилась к 30—40 гг. XX в., насчитывала немало интереснейших и довольно общих реакций, которые при ближайшем рассмотрении оказывались совершенно неудовлетворительными как рабочие инструменты для синтеза.
В этом отношении показательным примером является реакция Вюрца, открытая еще на заре развития органической химии (1855 г.). Первоначальный вариант этой реакции состоял в обработке алкилиодидов R-I натрием, в результате чего с неплохим выходом получались алканы R—R в соответствии с общей схемой:
R-I + R-I + 2Na → R-R + 2NaI
По-видимому, это сочетание явилось первым примером органической реакции, приводящей к образованию новой углерод-углеродной связи, и синтетическая ценность его как метода могла показаться бесспорной. Действительно, во многих учебниках по органической химии, начиная с элементарных курсов, и сейчас о реакции Вюрца говорится как о стандартном методе синтеза углеводородов. На практике же эта реакция применяется лишь при решении довольно узкого круга задач, в частности для синтеза циклических производных (подробнее об этом см. разд. 2.6.2.1.). Основная причина подобной «непопулярности» состоит в том, что в своем классическом варианте сочетание по Вюрцу оказывается не очень эффективным как препаративный метод, если его попытаться применить для сочетания двух разных алкилиодидов, R1—I и R2—I. Конечно, при этом будет образовываться требуемый продукт R1-R2, но лишь в смеси с продуктами симметричного сочетания R1—R1 и R2—R2. К тому же металлический натрий, применяемый в этой реакции в качестве конденсирующего реагента, настолько активен, что практически невозможно проводить эту конденсацию в тех случаях, когда субстрат содержит какие-либо дополнительные функциональные группы. В Дальнейшем мы покажем, каким именно образом удалось избавиться от указанных осложнений, а пока ограничимся только простой констатацией того факта, что именно неизбирательность протекания реакции являлась на протяжении многих десятилетий главным препятствием для широкого использования классической реакции Вюрца как метода в органическом синтезе.
Итак, как мы убедились, однозначность протекания реакции, ее «чистота», является еще одним и важнейшим критерием для оценки препаративной ее ценности. Этот важнейший момент заслуживает дополнительного обсуждения, снова на материале классической органической химии.
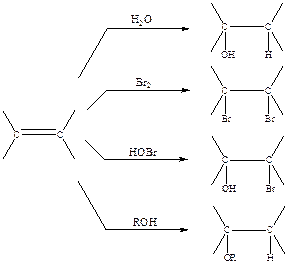 Схема 2.9
Схема 2.9
|
Способность алкенов присоединять такие реагенты, как вода, бром, органические и неорганические кислоты, спирты и т. п., давая при этом соответствующие аддукты (схема 2.9), была известна уже на раннем этапе развития органической химии.
Однако лишь немногие из показанных реакций оказались пригодными в классическом варианте их проведения в качестве методов синтеза целевых соединений, и здесь основным ограничивающим обстоятельством также явилась недостаточная чистота реакции. Действительно, даже в простейшем случае, например, гидратации этилена в присутствии серной кислоты, может образоваться помимо целевого продукта, этилового спирта, также диэтиловый эфир, диэтилсульфат и еще ряд продуктов (схема 2.10), и только при тщательной оптимизации условий реакции (кислотности среды, температуры, скорости подачи этилена, удаления продукта реакции и т. п.) можно обеспечить преимущественное образования одного из этих продуктов. Еще хуже обстоит дело в случае более сложных алкенов, так как при этом ход формально столь же простых реакций дополнительно осложняется возможностью образования продуктов присоединения как по правилу Марковникова (М-аддукт), так и против правила Марковникова (аМ-аддукт).
Конечно, в литературе можно найти немало примеров получения тех или ййыхсоединений из показанных на схеме 2.10 типов, но в большинстве этих случаев (за исключением, пожалуй, бромирования) требовалась детальная Отработка процедуры для каждого конкретного случая, и можно было лишь с d*teHb большой осторожностью говорить о применимости этой методики для субстратов более сложного строения.
Поэтому для того, чтобы стало возможным реальное синтетическое использование этих реакций, требовалось либо кардинально изменить условия проведения, либо разработать эквивалентные по результату синтетические последовательности, основанные на реагентах иной химической природы. Скажем, адекватной заменой кислотно-катализируемого присоединения воды или спиртов к алкенам в настоящее время является связка двух действительно общих и чисто протекающих (с исключительным образованием М-аддуктов!) реакций, а именно сольвомеркурирования и восстановления (см. схему 2.10).
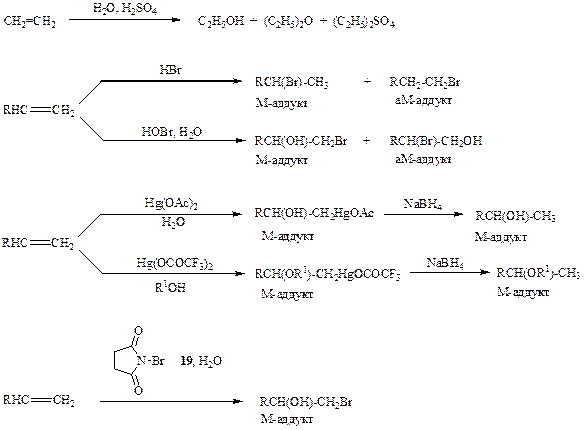 Схема 2.10
Схема 2.10
|
Конечно, внешне эта последовательность может показаться проигрышной по сравнению с прямым присоединением воды или спиртов, но ее отличительная особенность состоит в том, что эта методика применима практически для субстратов любого строения, что многократно подтверждено ее широким использованием в синтезе самых различных соединений.
Для чистого присоединения элементов бромноватистой кислоты (Вг+ и НО-) не потребовалось столь радикальной замены — в современном варианте достаточно гладкое образование соответствующих бромгидриноз может быть достигнуто при использовании N-бромсукцинимида (19) (как источника Вг+) в водной среде.
Итак, мы могли убедиться, что между реакциями, характеризующими свойства двойной связи как функциональной группы, и синтетическими методами, реализующими эти возможности, может действительно лежать «дистанция огромного размера».
Несколько неопределенное понятие «чистоты», которым мы выше пользовались, на самом деле представляет собой суммарную характеристику ряда независимых особенностей химического превращения. Среди них основными являются селективность реакции (этому важному аспекту далее будет посвящен разд. 2.4), отсутствие побочных реакций превращаемой функциональной группы и возможность проведения реакции в мягких условиях, которые исключают осложнения, связанные с наличием других функциональных групп в субстрате или лабильностью продукта реакции. Именно необходимость выполнения этих условий и является причиной того, что превращение, казалось бы, хорошо известной реакции в надежный синтетический метод обычно достигается ценой кропотливых и долгих поисков. Подобная работа может быть лишена внешних атрибутов новаторского творчества, однако в действительности только благодаря таким разработкам достигается реальное обогащение тактического арсенала органического синтеза. Поэтому справедливо считается, что заслуги создателей новых методов на основе даже известных реакций вполне соизмеримы с заслугами первооткрывателей этих реакций.
Ранее мы уже говорили о реакции Вюрца как потенциально полезном, но мало практичном (в классическом варианте!) методе создания связи С-С. Действительно, отмеченные выше ограничения области применения этого метода сохраняли свою силу на протяжении более чем ста лет после открытия базовой реакции. Однако в 60-х годах XX в. удалось разработать подходы, позволившие практически устранить недостатки, казалось бы, органически присущие сочетанию по схеме Вюрца. Посмотрим теперь, как же проходила эта разработка.
Первые попытки улучшить препаративную сторону реакции Вюрца базировались на расчленении суммарной реакции образования продукта сочетания на две, отдельно проводимые, стадии, а именно: превращение одного из галогенопроизводных в натрийорганическое соединение (стадия 1) и взаимодействие последнего со вторым компонентом (стадия 2, схема 2.11). Однако натрийорганические производные недостаточно устойчивы, и, кроме того, уже в момент своего образования они способны реагировать с исходным алкилгалогенидом, давая нежелательный продукт симметричного сочетания.
 Схема 2.11
Схема 2.11
|
Ситуацию удалось несколько улучшить при использовании магнийорганическихреагентов, но здесь на первый план выходит осложнение (общее и i ГдляNa- и для Mg-органических соединений) — возможность трансметаллирования,как это показано на схеме 2.11.
Поэтому настоящее решение проблемы создания работоспособного син-i 'тспсческого метода стало возможным лишь после введения в обиход нового поколения металлоорганических реагентов, а именно купратных реагентов — магний- или литийорганических соединений, модифицированных солями меди. Далее мы неоднократно будем встречаться с этими реагентами, а ' шока отметим, что их способность к трансметаллированию резко понижена, в силу чего достигается практически полная однозначность их взаимодействия с алкигалогенидами с образованием желаемого продукта сочетания R1—R2. К тому же купратные реагенты способны алкилироваться не только алкилгалогенидами, но и сложными эфирами спиртов (ацетатами, тозилата-ми), что, конечно, существенно расширяет возможности выбора реагентов, участников сочетания. Наконец, в отличие от соответствующих магний- или литийорганических соединений, купратные реагенты инертны по отношению к таким группам, как кетонная, что позволяет без осложнений реализовать сочетание по формальной схеме реакции Вюрца даже при наличии в субстратах такого рода рсакционноспособных функций, что было просто недостижимо в классическом варианте реакции. Поэтому неудивительно, что стадию образования связи С—С с использованием этого подхода можно встретить в синтезах самых разнообразных соединений.
Наконец, нужно подчеркнуть значение еще одного требования к хорошему синтетическому методу — глубокое понимание механизма базовой реакции. Подобное понимание, включающее надежное знание о природе элементарных стадий и интермедиатах, создает прочную теоретическую основу для уверенного предсказания результатов реакции в применении к новым субстратам и позволяет в случае необходимости направленным образом изменять параметры реакции с тем, чтобы добиться нужного результата. В этой связи стоит отметить, что именно глубокое изучение механизма реакции и послужило основой для успешной разработки современного варианта реализации сочетания, эквивалентного по результату формальной схеме синтеза Вюрца.
Выразительнейший пример, иллюстрирующий исключительную важность теоретических исследований для создания новых методов, можно найти в истории проблемы перициклических реакций*.
 Схема 2.12
Схема 2.12
|
К реакциям этого класса, помимо уже рассмотренной выше реакции Дильса—Альдера ([4 + 2|-циклоприсоединение), относятся также реакции, описываемые формальной схемой циклодимеризации алкенов ([2 + 2]-цик-лоприсоединение, схема 2.12). С давних пор было известно множество разрозненных и не поддающихся рациональной трактовке случаев образования циклобутановых производных, протекающая только при нагревании или только при облучении, проявляющих различную чувствительность к вариациям структуры исходных веществ и подчас резко различных fci, йЬсгереохимическому результату.
На протяжении довольно длительного времени подобные реакции даже в серьезных монографиях рассматривались как «реакции без механизма» (no-mechanism reactions), т.е как процессы, для которых принципиально невозможно написать единый корректный механизм. Понятно, что подобная необъяснимость хода и непредсказуемость результатов делали почти невоз-fi ножным использование превращений этого типа для целей направленного ■ ринтеза. Эта ситуация кардинально изменилась в начале 1960-х годов, когда t Р. Вудвордом и Р. Хоффманом была создана теория перициклических реак-> рий, которая позволила обобщить и интерпретировать с единой точки зре-f rim многочисленный и разнородный экспериментальный материал [2а]. Не Лсаясь основного содержания теории Вудворда—Хоффмана (соответствую-щкй материал можно найти в любом современном учебнике по органиче-dfflt химии), отметим, что в этой теории выделяется два крайних типа Механизмов циклоприсоединения: один для реакций, инициируемых терми-Й, т. е. не требующих ни катализаторов, ни облучения (такова, напри-р, реакция Дильса—Альдера), и другой для реакций, инициируемых облу-чйгием. В превращениях первого типа реагирующие соединения находятся в основном состоянии, в то время как реакции второго типа осуществимы ffitin, в возбужденном состоянии. Каждый из этих типов реакций характерен сЬоими особенностями стереохимии и осуществим лишь для субстратов со-ВЙршенно определенного типа строения. В применении к циклодимериза-iflfat алкенов, показанной на схеме 2.12, теоретический анализ показал, что фотохимическая реакция относится к категории перициклических реакций [2 + 2]-циклоприсоединения, в то время как термическая циклизация проте-*ает по совершенно иному, чаще всего ступенчатому механизму. Эти представления позволили с единой точки зрения рассмотреть и обобщить уже накопленный, подчас очень противоречивый экспериментальный материал (так сказать, «навести порядок в хозяйстве») и четко определить особенности Направленности, оптимальные условия и области применимости обеих реакций. Именно благодаря достигнутому глубокому пониманию механизма уда-Лось разработать серию методов, превративших циклодимеризацию алкенов в один из наиболее надежных и мощных инструментов современного органического синтеза, уникальный синтетический потенциал которого сделали возможным построение множества полициклических соединений с самым причудливым углеродным скелетом (подробнее об этом см. разд. 2.6.3).
В заключение этого раздела нам бы хотелось еще раз обратить внимание читателя на следующее обстоятельство: ни один даже самый изощренный современный метод не может являться универсальным, так сказать, абсолютным методом решения данной тактической задачи. Любой метод ограничен по своей применимости и не только спецификой конкретной структуры субстрата, но также и тем, что необходимые для его использования условия могут инициировать протекание разного рода побочных превращений как исходных реагентов, так и конечных продуктов. Именно в силу этих обстоятельств на самом деле необходима кажущаяся бесконечной работа по созданию новых и модификации старых методов проведения даже хорошо изученных реакций.
Рассмотрим теперь более конкретно основные синтетические методы, и прежде всего методы построения углеродного скелета молекул.
– Конец работы –
Эта тема принадлежит разделу:
ОРГАНИЧЕСКИЙ СИНТЕЗ
ORGANIC SYNTHESIS... THE SCIENCE BEHIND THE ART...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Органическая реакция и синтетический метод
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.031 сек.
Новости и инфо для студентов