рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Образование
- /
- Альтернативных реакционных центров субстрата
Реферат Курсовая Конспект
Альтернативных реакционных центров субстрата
Альтернативных реакционных центров субстрата - раздел Образование, ОРГАНИЧЕСКИЙ СИНТЕЗ Классический Пример Такого Подхода К Решению Проблемы — Ацетоуксус-Ный Эфир ...
Классический пример такого подхода к решению проблемы — ацетоуксус-ный эфир (168).Его обычной реакционноспособной формой является 1енолят 169,реакции которого с разнообразными С-нуклеофилами протекают по центральному атому углерода. Последующий гидролиз продукта 170и декарбоксилирование приводят к образованию кетона 171.Нетрудно видеть, что структура последнего соответствует продукту взаимодействия того же электрофила с енолятом ацетона 172,и, следовательно, в показанной на схеме 2.80 последовательности реакций енолят ацетоук-сусного эфира 169на самом деле используется в роли эквивалента енолята 172.
 Схема 2.80
Схема 2.80
|
Зададимся теперь вопросом: а зачем, собственно, здесь нужна подобного рода замена простого на более сложное? Ведь хорошо известно, что енолят 172достаточно легко может быть получен из ацетона при действии оснований, и алкилирование 172также само по себе не составляет какой-либо проблемы. Однако следует учитывать, что ацетон сам является достаточно активным электрофилом, и поэтому трудно избежать его реакции с образующимся енолятом 172(альдольная конденсация, см. разд. 2.2.3.2). Но тут есть и второе осложнение, более существенное в свете обсуждаемой проблемы селективности. Дело в том, что в получающихся продуктах алкилирования 171также содержится функциональная группа СО-Ме, мало отличающаяся по своим свойствам от подобной функциональной группы исходного ацетона.
Поэтому в условиях реакции будет также происходит депротонирование ке-тона 171с последующим алкилированием образовавшегося енолята электро-филом. Отсюда следует, что прямой путь получения 171непосредственно из ацетона в общем случае малопригоден из-за присущей ему низкой селективности образования целевого продукта.
Иное дело ацетоуксусный эфир 168.В молекуле этого соединения также имеются две группы, способные к ионизации. Однако благодаря эффекту двух электроноакцепторных групп кислотность протонов СН2-фрагмента на несколько порядков выше, чем кислотность протонов метальной группы. По этой же причине енолят 169гораздо стабильнее енолята 173.В силу этих обстоятельств даже под действием сравнительно слабых оснований (например, спиртового раствора алкоголята натрия) 168 практичеки нацело превращается в енолят 169,чем и обеспечивается селективность образования продукта алкилирования 170.Таким образом, временно введя в молекулу ацетона карбэтоксильную группу, т.е. перейдя к ацетоуксусному эфиру, мы добились резкой активации одной из метальных групп (превращенной таким способом в метиленовую) и тем самым обеспечили селективность реакций с электрофилом. Именно на этом принципе и базировалось использование ацетоуксусного эфира в органическом синтезе на протяжении многих десятков лет.
Но это еще не все — потенциал ацетоуксусного эфира, классики не только синтетической, но и теоретической органической химии, далеко не исчерпывается рассмотренными выше превращениями. Оказалось, что, следуя тем же принципам, можно, как мы сейчас увидим, добиться обратной селективности алкилирования по альтернативным положениям и провести эту реакцию исключительно по метильной группе. Идея такого рода может показаться парадоксальной, но на самом деле ее реализация выглядит достаточно логично. Так, если генерировать енолят 169в апротонной среде и далее обработать его еще 1 экв. более сильного основания (например, бутил-лития или диизопропиламида лития, LDA), то происходит повторная ионизация, приводящая к образованию бис-аниона 174(схема 2.81).
 Схема 2.81
Схема 2.81
|
Наличие двух отрицательных зарядов делает интермедиат 174чрезвычайно активным. Однако имеющиеся в нем карбанионные центры при С-1 и С-3 достаточно сильно различаются по своей реакционной способности. Благодаря наличию двух карбонильных заместителей карбанионный центр нри С-3 гораздо более стабилизирован по сравнению с центром при С-1. Следовательно, именно последний должен легче подвергаться атаке элект-дофилом, и, действительно, результатом реакции бис-аниона 174с одним аквивалентом какого-либо электрофила является исключительное образование продукта атаки по этому центру 175.Поскольку последний также содержит снолятный фрагмент, то возможно провести еще одну стадию алкилирования тем же или иным электрофилом, на этот раз по центру С-3. Таким образом, ацетоуксусный эфир, в лице своего бис-аниона 174может использоваться как С3-блок для синтеза самых различных кетонов типа 176,а если нужно, то может также служить и С»-блоком для построения эфиров кето-кислот 177, структура которых задается природой и порядком прибавления реагентов Е1 и Е2 [24т].
Двухзарядные или даже трехзарядные органические ионы, структура которых обеспечивает одновременно высокую региоселективность и реакционную способность, сравнительно недавно вошли в обиход синтетической практики и широко используются в настоящее время. К ним относятся, помимо дианиона 174,такие производные, как дианионы карбоновых кислот J78,пропаргильный дианион 179 и дианион пропаргилового спирта 180(схема 2.82). Селективность электрофильной атаки для этих интермедматов также определяется относительной нуклеофилъноетъю анионных центров, которая обратна их термодинамической стабильности: менее стабилизированный центр является предпочтительным местом атаки (на схеме 2.82 эти центры обозначены звездочкой).
 Схема 2.82
Схема 2.82
|
Применение подобных полианионов позволило существенно расширить синтетические возможности химии карбанионных реагентов. Уместно подчеркнуть, что это стало возможным именно благодаря разработке методов, основанных на использовании в качестве универсальных реагентов для генерации карбанионных интермедиатов сильных ненуклеофильных оснований типа LDA.
На примере ацетона и ацетоуксусного эфира мы рассмотрели простейший пример того, каким образом может решаться задача селективного алкилирования только одного из двух идентичных а-положений. Классическое решение такого рода задач для кетонов всевозможного строения долгoe время строилось на аналогичной основе путем искусственного создания в требуемом месте группировки типа ацстоуксусного эфира, например, по реакции енолята с хлоругольным эфиром C1COOR. Нетрудно, однако, видеть, что такой подход применим лишь в случае симметричных кетонов, где два енолята идентичны, а последующее ацилирование приводит к идентичным продуктам. В случае же несимметричных кетонов использовать принцип такого рода селективной активации довольно затруднительно, в первую очередь из-за малой селективности стадии генерации енолятов.
Эти и другие осложнения заставили в 1960-х годах предпринять углубленные исследования в области химии енолятов, в результате чего удалось разработать ряд приемов, обеспечивающих надежное решение проблемы селективности генерации енольных производных для карбонильных соединений самого различного строения.
Первоначально найденное решение, хотя и было достаточно общим и надежным, оказалось все-таки не очень удобным. Смысл его сводился к следующему: смесь енолятов, образующихся при взаимодействии несимметричных кетонов, например 1-метилциклогексанона (181),с такими основаниями, как триэтиламин в среде апротонного растворителя, обрабатывали триметилхлорсиланом — электрофилом, атакующим еноляты исключительно по атому кислорода. Получаемую при этом смесь региоизомерных триме-тилсилиленолятов (например, 182аи 182Ь,схема 2.83) разделяли перегонкой, после чего из индивидуальных изомеров действием метиллития получали in situ соответствующие литиевые еноляты 181аи 181Ь,взаимодействие которых с требуемым электрофилом приводило к получению чистых региоизоме-ров 183аи 183Ь.
Входе дальнейших исследований было замечено, что изомерный состав смеси силильных енолятов зависит от «предыстории» реакции: если триме-тилхлорсилан прибавляют в реакционную смесь немедленно после введения основания, то образуется преимущественно изомер 182а,а если полученную на стадии енолизации смесь литиевых енолятов выдержать некоторое время в отсутствие электрофила, то ее последующее «гашение» триметилхлорсиланом приводит к смеси силиловых эфиров енолов другого изомерного состава (с преобладанием 182Ь)[25а]. Отсюда следовало, что первоначальным результатом енолизации является отрыв протона от наименее затрудненного из а-атомов углерода кетона 181с образованием енолята 181а,являющегося таким образом кинетическим продуктом, который и может быть «зафиксирован» в виде силильного производного 182а,если в среде присутствует триме-тилхлорсилан. При отсутствии последнего может происходит обратимая изомеризация 181ав 181Ь,и от соотношения изомеров вравновесной смеси и зависит состав смеси силиловых эфиров 182аи 182Ь,получаемых при последующем добавлении триметилхлорсилана. Преобладание 182Ьв этой смеси означает, что 181Ьявляется термодинамически более стабильным изомером, так что образование 182Ьпроисходит за счет термодинамически контролируемого процесса.
 Схема 2.83
Схема 2.83
|
На основании этих данных удалось разработать методики, который позволяли с высокой селективностью получать силиловые эфиры как кинетически контролируемыхенолятов, так и их термодинамически более стабильных изомеров. Так, если проводить енолизацию кетона 181 с помощью сильного и сте-рически затрудненного основания, такого, как бис-триметилсилиламида лития (BSA) при возможно более низкой температуре с немедленной обработкой реакционной смеси триметилхлорсиланом, то енольный эфир 182а получается с чистотой более 90%. Если же использовать для енолизации более слабое основание, триэтиламин, и проводить реакцию при нагревании, то с такой же чистотой можно получить изомерный продукт 182Ь (см. схему 2.83) [25Ь].
Принцип такого подхода оказался достаточно общим и на его основе разработано множество вариантов региоселективного получения коваленгных енольных производных не только кремния, но и других элементов, например, олова, титана, циркония и бора, которые широко применяются в современном синтезе как эквиваленты ионных енолятов.
Заслуживает упоминания еще один немаловажный аспект химии силило-вых эфиров енолов. Как мы уже отмечали, реакции этих ковалентных производных с электрофилами требуют присутствия кислот Льюиса (см. схемы 2.41—2.44). Существует, однако, альтернативный путь инициирования этой реакции, а именно под действием соли //-ButN+F", которая служит источником несольватированного фторид-иона в среде апротоных органических растворителей [17с]. Такое специфическое действие фтор-аниона обусловлено тем, что коваленгная связь Si—F принадлежит к числу самых прочных одинарных связей и ее образование — высоко экзотермический процесс. Результатом реакции фторид-иона с силиловыми эфирами енолов является расщепление связи Si—О и образование соответствующего тетрабутиламмониевого енолята, который, как и полагается ионным енолятам, легко взаимодействует с ковалентными электрофилами самой различной природы (схема 2.84).
 Схема 2.84
Схема 2.84
|
Итак, общим методом избирательной активации альтернативных положений в несимметричных кетонах может служить селективная енолизация с образованием ковалентных производных, результатом которой является по сути дела создание нуклеофильного центра при одном или другом из а-угле-родных атомов.
Среди других способов, также позволяющих решать задачу селективности алкилирования кетонов, следует упомянуть методы, основанные на использовании в качестве субстратов некоторых производных кетонов, например иминов, гидразонов или оксимов [4]. В этих методах региоселективность образования ионных енолятов определяется действием таких факторов, как способность атома азота образовывать координационную связь с катионом лития и стерическими препятствиями, создаваемыми заместителями при атоме азота. Результатом этих эффектов является высокая предпочтительность енолизации по менее замешенному а-атому, что особенно явно выра-жено для диметилгидразонов кетонов [25с]. На этой основе разработан ряд якнтетически интересных превращений, в которых осуществляется контролируемое последовательное алкилирование исходного субстрата по обоим его а-атомам. Пример подобной последовательности реакций, проводимых Людном реакционном сосуде и состоящей из двух последовательных цик-лов — енолизация + алкилирование, показан на схеме 2.85 [25d].
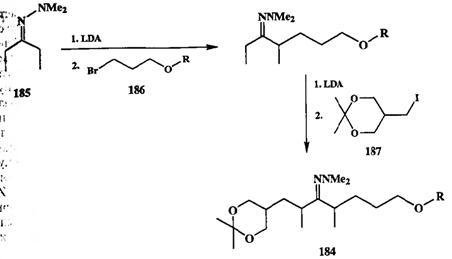 Схема 2.85
Схема 2.85
|
– Конец работы –
Эта тема принадлежит разделу:
ОРГАНИЧЕСКИЙ СИНТЕЗ
ORGANIC SYNTHESIS... THE SCIENCE BEHIND THE ART...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Альтернативных реакционных центров субстрата
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.028 сек.
Новости и инфо для студентов