рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Образование
- /
- Циклоприсоединение
Реферат Курсовая Конспект
Циклоприсоединение
Циклоприсоединение - раздел Образование, ОРГАНИЧЕСКИЙ СИНТЕЗ Среди Множества Реакций, Относящихся К Этому Классу, Особое Место Занимает [...
Среди множества реакций, относящихся к этому классу, особое место занимает [4 + 2]-циклоприсоединение. Это — реакция Дильса—Альдера [2а], как правило, не требующая катализа или иницирования облучением. В этой реакции происходит образование шестичленного цикла в результате взаимодействия сопряженного диена ^-компоненты) и диенофила ((^-компоненты) с образованием циклического переходного состояния, в котором шесть л-электро-нов исходных соединений образуют единое электронное облако квазиароматического типа (324, схема 2.117) [31а,B].
Именно благодаря такому эффекту стабилизации переходного состояния последнее оказывается достаточно энергетически выгодным, а потому энергия активации реакции относительно низкой. Для несимметрично замещенных диенов и диенофилов возможно образование более чем одного переходного состояния типа 324. Однако эти изомерные переходные состояния достаточно различны по энергии, вследствие чего наиболее обычным результатом реакции Дильса—Альдера является исключительное, или по крайней мере преимущественное, образование одного из возможных изомерных продуктов (по положению или взаимной ориентации заместителей). Ход реакции адекватно объясняется в рамках концепции сохранения орбитальной симметрии Вудворда—Хоффмана [31с], и, как правило, конечный результат реакции хорошо предсказуем даже для очень непростых случаев [31d].
 EWG – электроноакцепторная группа
Схема 2.117
EWG – электроноакцепторная группа
Схема 2.117
|
В классическом варианте реакции Дильса—Альдера в качестве 4л-компо-ненты используются различные соединения, содержащие 1,3-диеновый фрагмент, а в качестве 2я-компоненты, диенофила, — алкены или алкины, содержащие электроноакцепторные группы (EWG), такие, как сопряженные альдегиды, кетоны, кислоты и их производные, нитроалкены и т. д. Типичный набор карбо- и гетероциклических структур, получаемых с помощью этой реакции, показан на схеме 2.118.
 Схема 2.118
Схема 2.118
|
Для того чтобы более полно охарактеризовать достоинства реакции Дильса—Альдера как препаративного метода, полезно хотя бы вкратце остановиться на особенностях регио- и стереонаправленности этой реакции. Прежде всего, следует отметить, что сам механизм реакции подразумевает, что образование циклических продуктов протекает с полным сохранением'конфигурации заместителей в обоих фрагментах (см. превращения 325 ->• 326 или 327 -> 328 на схеме 2.119). Для несимметричных диенов и диенофилов наблюдается преимущественное образование одного из возможных региоизо-меров (см., например, образование 329 или 330). Для 1,4-дизамещенных диенов возможно образование двух переходных состояний, отвечающих экзо-или энйо-ориентации рееагентов. Типичным результатом циклоприсоедине-ния в таких случаях является преимущественное или даже исключительное образование эядо-аддуктов (см., например, 331а).
В рамках общей схемы [4 + 2]-циклоприсоединения можно реализовать синтезы с введением в состав собираемого шестичленного фрагмента самых неожиданных (на первый взгляд!) заместителей, что может быть обеспечено подбором соответствующих структур 4л- или 2я-компоненты с тем, чтобы они содержали легко трансформируемые или удаляемые группы.
Рассмотрим, например, ретросинтетический анализ структур 332 или 333 на схеме 2.120. Для того чтобы представить себе возможность сборки таких продуктов по схеме диенового синтеза, полезно прежде всего превратить их (ретросинтетически) в енолы 332а и 333а соответственно. Если далее применить разборку по схеме ретродиенового синтеза, то мы автоматически придем к структурам 334 и 335 для диеновых компонент.
Может показаться, что такой путь разборки неконструктивен, так как подобного рода диены неспособны существовать как реагенты. Однако все становится на свои места, если вспомнить, что разнообразные силиловые эфиры енолов относятся к категории вполне доступных и стабильных производных. Понятно, что с помощью диенов типа силиловых эфиров 336 и 337, используемых в качестве стабильных эквивалентов диенов 334 и 335, требуемые превращения легко могут быть осуществлены [31е] (см. схему 2.121).
Другие примеры, иллюстрирующие некоторые неочевидные возможности Применения диенового синтеза с участием модифицированных диенов и диенофилов, приведены на схеме 2.122. В синтезе природного сесквитерпена гидроксиденталола (338а) ключевая стадии построения бициклического скелета, содержащего 1,3-диеновый фрагмент (продукт 338), выполняется по схеме диенового синтеза с использованием пирона-2 (339) как диеновой компоненты и диенофила 340.Первоначально получаемый при этом аддукт 340а легко подвергается декарбоксилированию в условиях реакции, что и приводит к получению требуемого промежуточного продукта 338 [31П. Использование как диена 338,так и производных, содержащих в своем составе подобного рода фрагмент, достаточно распространено в синтезе самых различных полициклических соединений, содержащих циклогексадиеновый остаток [31g]. Силоксидиен 341был сконструирован для того, чтобы обеспечить возможность получения циклогексадиеновых и ароматических аддуктов с необычным типом замещения. Как показано на схеме 2.122, диеновый синтез с участием 341и ацетиленового диенофила дает аддукт 342.Кислотный гидролиз последнего приводит в конечном счете к тетразамещенному ароматическому
 Схема 2.119
Схема 2.119
|
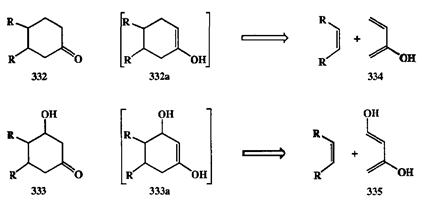 Схема 2.120
Схема 2.120
|
продукту 343,получение которого другим путем было бы довольно затруднительно. Продукт 343далее использовался в синтезе одного из природных ингибиторов роста растений, лазиодиплодина [31h].
Интересен синтетический потенциал, открываемый при проведении диенового синтеза с диеном 344.Непосредственно образуемые при этом аддук-ты типа 345содержат группировку аллилсилана, что позволяет вводить их в реакции с различными электрофилами. Результатом последовательности превращений — диеновый синтез плюс электрофильное присоединение — является образование продуктов типа 346(3 li] (схема 2.122), структура которых сама по себе никакие предполагает возможности их синтеза по схеме реакции Дильса—Альдера.
Сказанное выше в полной мере относится и к примерам вариаций на тему природы диенофила, показанным на схеме 2.122а. Так, диенофил 347позволяет синтезировать (по реакции с циклопентадиеном) аддукт 348,из которого легко получаются аддукты 349 или 350 [31j]. В принципе, как 349, так к 350 могут быть получены непосредственным взаимодействием циклопентадиена с этиленом или соответствующим терминальным алкеном, однако условия подобных превращений с участием неактивированных диенофилов настолько жестки, что в препаративном плане такие возможности особого интереса не представляют. Напротив, благодаря использованию в качестве диенофила сульфона 347 как синтетического эквивалента алкеновых синтонов 347а и 347Ь, стало возможным применять реакцию Дильса—Альдера как общий метод синтеза циклических аддуктов, не содержащих электроноакцепторных групп.
 Схема 2.121
Схема 2.121
 Схема 2.122
Схема 2.122
|
Наличие трифенилфосфониевого фрагмента делает винильное производное 351 активным диенофилом. Легко видеть, что получаемая в результате реакции 351 с бутадиеном циклическая фосфониевая соль 352 представляет собой готовый реагент для генерации соответствующего фосфорана и синтеза (по Виттигу) диенов с экзоциклической двойной связью типа 353 [31k]. Получение последних непосредственно по схеме диенового синтеза потребовало бы использования в качестве диенофила аллена RCH=C=CH2, что мало перспективно из-за термической лабильности алленов.
 Схема 2.122а
Схема 2.122а
|
Наконец, последний пример, показанный на схеме 2.122а, иллюстрирует возможность использования 1,1-дизамещенных диенофилов, например 354, как эквивалентов кетена. Действительно, аддукт 355, получаемый в результа-те взаимодействия замещенного циклопентадиена с диенофилом 354, может далее подвергаться щелочному гидролизу, что и приводит к кетону 356 [311], ключевому полупродукту в одном из первых стереоселективных синтезов простагландинов. Отметим, что 356 является продуктом формального диенового синтеза с участием кетена в качестве диенофила, однако из-за низкой активности (и малой термической стабилькости) кетена непосредственно реализовать такой синтез не удается.
Совершенно особое место занимает реакция Дильса—Альдера в синтезах полицикических структур каркасного типа, таких, как, например, баскетен (357) (схема 2.123) [31m]. В этом случае реакция между диеном 358 (в этой форме реагирует циклоктатетраен) и малеиновым ангидридом позволяет сразу получить с количественным выходом трициклическую структуру 359.Последующая стадия [2+2]-циклоприсоединения (об этой реакции см. следующий раздел) приводит к образовнию каркасного соединения 360,из которого в результате последовательности несложных стадий (омыление и окислительное декарбоксилирование) был получен целевой продукт 357.
Столь же эффективно используется диеновый синтез на ключевых стадиях получения кубана (361),пентапризмана (362)[31п] и многих других представителей этого экзотического класса органических соединений. Можно смело утверждать, что вообще синтетики вряд ли бы предпринимали синтез структур такой степени сложности, если бы не располагали столь мощным методом, как реакция Дильса— Альдера.
Начиная с 1960-х годов все более важную роль в полном синтезе начинает играть внутримолекулярная реакция Дильса—Альдера [31о]. На схеме 2.124 приведена выборка некоторых представительных примеров, позволяющая судить об особенностях протекания внутримолекулярного варианта диенового синтеза и специфике его использования в полном синтезе.
Как мы уже отмечали, алкены, не содержащие электроноакцелторных групп, являются очень «вялыми» диенофилами, и случаи их препаративного использования в межмолекулярном диеновом синтезе довольно редки. Показанный на схеме эффективный синтез крайне напряженной структуры брексена (363)в одну стадию из доступного предшественника 364(через стадию равновесной изомеризации последнего в 364а)по схеме внутримолекулярного диенового синтеза наглядно показывает уникальные препаративные возможности этого метода [31р].
 Схема 2.123
Схема 2.123
|
Превращения 365а -* 366аи 365Ь -> 366Ъ[3 lo,r] приведены как типичные примеры, иллюстрирующие особенности структурной и пространственной направленности реакции. Отметим, в частности, что региоселективность образования этих аддуктов обратна той, которую можно было бы ожидать на основании сравнения с аналогичным превращениям в межмолекулярном варинте 367+ 368-> 369.Интересно также что, если образование 366алегко описывается в терминах стандартной схемы эидо-присоедине ния, то для интерпретации стереохимического результата превращения 365Ь -* 366Ьнеобходимо предположить исключительную экзо-ориентацию реагирующих фрагментов. Возможность реализации схем как эндо-, так и экзо-присоединения может показаться серьезным недостатком внутримолекулярной реакции Дильса—Альдера, однако на самом деле обычно бывает нетрудно предсказать результат для того или иного конкретного примера на основе рассмотрения особенностей несвязанных взаимодействий в альтернативных структурах соответствующих переходных состояний. Более того, удается также направлять реакцию по пути экзо-или эидо-присоединения подбором размера цепи, связывающей реагирующие центры и/или введением каких-либо вспомогательных групп [31 о].
Внутримолекулярное [4 + 2] -циклоприсоединение было использовано на ключевой стадии синтеза форскиолина (370)[31s] (схема 2.124), природного биологически активного вещества. Для получения требуемого предшествен ника легко доступный спирт 371 был превращен в эфир 372. Последний содержит в своем составе и диенофильную группу, и диеновый фрагмент. Хотя диены подобной степени замещенности обычно бывают крайне инертными компонентами в межмолекулярном варианте диенового синтеза, внутримолекулярная циклизация 372 -> 373 протекала достаточно легко и дала требуемый продукт с почти количественным выходом. Характер функциональности аддукта 373 позволил далее относительно легко завершить построение структуры 370.
Очевидные преимущества внутримолекулярной реакции Дильса—Альде-ра стимулировали поиски с целью разработки способов создания временных мостиков между диеном и диенофилом. Особенно плодотворными оказались исследования в этой области, выполненные в группе Сторка [31t], целью которых было выяснение возможностей использования в качестве мо-стиковых групп фрагментов, содержащих атомы кремния, магния или алюминия. Эффективность такого подхода показана на примере разных вариантов модельного превращения 374 -> 375 через промежуточные стадии 374а и 374Ь (схема 2.124). Примечательно, что диеновый синтез протекал особенно легко в том случае, когда в качестве мостиковых гетероатомов использовалась пара O-Mg (374a, Z=Mg). Подкупающе проста процедура получения подобного рода производных взаимодействием литиевого алкоголята спирта 374 с винилмагнийбромидом. Хотя диенофилом в структуре 374а является по сути дела карбанионный фрагмент, тем не менее превращение 374а (Z=Mg) -> 374b протекало в довольно мягких условиях (нагревание при 80°С в течении нескольких часов). Та же реакция другого, явно ковалентного производного — кремнийсодержащего аналога 374а (Z=SiMe2) — требовала нагревания при 160°С. Отметим, что для всех производных типа 374Ь завершающая стадия — превращение в продукт 375 — легко достигалось путем гидролиза. Примечательная эффективность внутримолекулярного диенового синтеза побудила более внимательно изучить вопросы биогенеза некоторых типов полициклических соединений, синтез которых по этой схеме казался вполне реальным с точки зрения синтетиков. Оказалось, что Природа не менее нашего «осведомлена» о подобных возможностях, и действительно для ряда примеров было экспериментально доказано использование внутримолекулярной циклизции по Дильсу—Альдеру как одной из стадий биосинтеза. Так, из патогенных грибов Alternaria solan's были выделены фитотоксины, со-ланопироны А и D (376а и 376Ь). Сам факт выделения пары диастереомеров из одного и того же природного источника был достаточно необычным, ибо в подавляющем большинстве случаев биосинтетические превращения протекают с абсолютной стереоселективностью. Изучение биосинтеза этих соединений с использованием изотопно меченых соединений показало, что они образуются в результате циклизации общего предшественника 377 [31u,v], как это показано на схеме 2.125.
 Схема 2.124
Схема 2.124
|
 Схема 2.125
Схема 2.125
|
Та же циклизация триена 377 при проведении ее в колбе (in vitro) протекала при нагревании и привела к образованию тех же диастереомеров 376а и 376Ь примерно в том же соотношении, в каком они образуются в клетке (in vivo) [31w]. Рассмотрение моделей показало, что циклизация 377 может проходить как по схеме эндо-присоединения (для конформера 377а), так и по схеме экзо-присоединения (для конформера 377Ь). Расчеты также показали, что эти конформеры, как и образующиеся из них переходные состояния циклизации, очень мало различаются по своей энергии. По-видимому, именно благодаря этим факторам циклизация 377 как in vivo, так и in vitro протекает с низкой стереоселективностью.
В последующих разделах мы еще неоднократно будем рассматривать примеры применения самых различных вариантов реакций Дильса—Альдера в синтезе.
– Конец работы –
Эта тема принадлежит разделу:
ОРГАНИЧЕСКИЙ СИНТЕЗ
ORGANIC SYNTHESIS... THE SCIENCE BEHIND THE ART...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Циклоприсоединение
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.027 сек.
Новости и инфо для студентов