рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Образование
- /
- Органические ионы и факторы, определяющие их стабильность
Реферат Курсовая Конспект
Органические ионы и факторы, определяющие их стабильность
Органические ионы и факторы, определяющие их стабильность - раздел Образование, ОРГАНИЧЕСКИЙ СИНТЕЗ Высокая Химическая Активность Карбокатионов И Карбанионов Свя...
Высокая химическая активность карбокатионов и карбанионов связана прежде всего с силами кулоновского взаимодействия. Точечный заряд, сосредоточенный на атоме углерода, создает электростатическое поле, оказывающее весьма сильное воздействие на все ближайшее окружение такого заряда. Прежде всего, это сильное притяжение к ионам противоположного знака — притяжение, которое может приводить к образованию ковалентной связи. Например, практически невозможно найти условия, которые бы обеспечили существование такой пары ионов, как метил-катион (23) и анион хлора, т.е. каким-либо образом предотвратить их немедленную ассоциацию с образованием ковалентного соединения — метилхлорида (24)*.
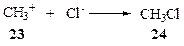
Далее, воздействие заряда иона на неионные молекулы усиливает поляризацию их связей и создает предпосылки для полной ионизации, т.е. в конечном счете для реакции между ионом и полярным соединением. Эти процессы составляют главную причину высокой и разнообразной реакционной способности ионов, причем, конечно, как желательной, так и нежелательной. В частности, и это очень существенная для органического синтеза частность, они резко ограничивают набор растворителей, инертных по отношению к этим частицам, а также и выбор функциональных групп субстратов, выдерживающих такое воздействие. Например, упоминавшийся выше ион ацетилия нельзя было бы удержать в спиртовом растворе или использовать для ацетилиро-вания бензилового спирта (25) по углероду ароматического кольца (реакция Фриделя—Крафтса) из-за мгновенно протекающих реакций:
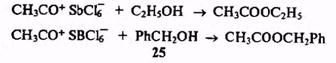
Поляризация связей, в том числе сопровождающаяся химическими реакциями, происходит не только в молекулах, окружающих ион, но также, и в первую очередь, в самих органических ионах. Так, сравнительно устойчивый ион, лу>етл-бутил-катион (26), в присутствии даже слабых оснований легко теряет протон с образованием изобутилена (27) (элиминирование, схема 2.14). При этом электронная пара, образующая связь С-Н, целиком «перетягивается» к положительному заряду, происходит выброс протона, уносящего положительный заряд, и образуется связь С=С.
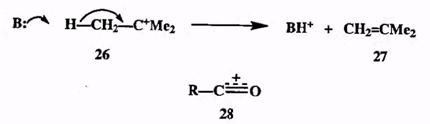 Схема 2.14
Схема 2.14
|
Из сказанного следует, что для того, чтобы как-то понизить реакционную способность органических ионов, подавить их склонность реагировать «без разбора с чем угодно», требуется в первую очередь ослабить их электростатическое взаимодействие с другими молекулами. Для этого имеются по крайней мере три возможности. Одна из них, наиболее общая, заключается в том, чтобы модифицировать структуру иона введением полярных или легко поляризуемых групп с тем, чтобы превратить точечный заряд, локализованный на одном атоме, в «размазанный», делокализованный на нескольких атомах. Полный заряд иона при этом, естественно, не меняется, но резко возрастает его эффективный радиус (а электростатические силы, как известно, обратно пропорциональны квадрату расстояния). Понятно, что подобная стабилизация для карбокатионов и карбанионов должна обеспечиваться разными по характеру группами. В случае карбокатионов это должны быть группы, способныеные подавать электроны к катионному центру (электронодонорные груп-пы, а в случае карбанионов — группы, способные оттягивать электроны от анионного центра (электроноакцепторные группы). Так, например, благодаря поляризации связей С—Н заряд в трет-буталъном катионе (26) распределен между центральным атомом углерода и девятью атомами водорода, т. е. в значительной мере делокализован. Благодаря такому эффекту этот катион в подходящих средах и при умеренно низкой температуре достаточно устойчив, хотя и сохраняет весьма высокую реакционную способность. B ацилий-ионах, отдельные представители которых настолько стабиль-ны, что могут быть получены в виде устойчивых солей в свободном состоя-нии, основная стабилизация достигается за счет частичного перетягивания неподеленной пары электронов кислорода к положительно заряженному углероду,так что изображать их правильнее в виде структур типа 28 с частичной тройной связью между атомами кислорода и углерода и распределе-нием заряда по обоим центрам. Сходным образом достигается стабилиза-циякарбокатионов за счет некоторых других гетероатомов, находящихся непосредственно у катионного центра или даже на некотором от него уда-лении[3].
Классическим примером стабильных карбанионов являются енолят-ионы, которые образуются при действии оснований на карбонильные соединения , содержащие хотя бы один атом водорода в а-положении. Такие ионы можнопредставить себе как резонансный гибрид предельных структур 29с зарядом на углероде) и 30 (с зарядом на кислороде) или, более лаконично, эквивалентной формулой 31, в которой штриховые связи и символ заряда означают, что электронная пара распределена между тремя атомами, а заряд — между двумя крайними атомами этой триады (схема 2.15).
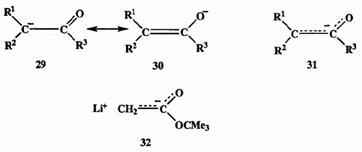 Схема 2.15
Схема 2.15
|
По сходному принципу осуществляется стабилизация карбанионов и другими электроноакцепторными группами, содержащими кратные связи, во фрагментах типа -C-CN, -C-COOR, -C-NO2 и т. п. [4]. Показанная на схеме литиевая соль трет-бутилацетата (32), которая может быть легко получена и стабильна при хранении, является наглядным примером эффективности такой стабилизации. Заместители, содержащие многоэлектронные лег-кополяризуемые гетероатомы типа серы или селена, также способны обеспечивать стабилизацию соседнего карбанионного центра.
Универсальным и эффективным «буфером» любого заряда, своего рода молекулярным конденсатором является ароматическое ядро. Его замкнутая система я-электронов способна легко смещаться и к заряду (положительному) и от заряда (отрицательного), т. е. легко поляризуется, что и приводит к делокализации заряда. Благодаря этому эффекту как бензил-катион (33), так и бензил-анион (34) оказываются относительно стабильными частицами.
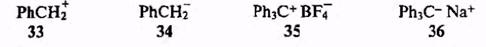
Вклады нескольких поляризуемых групп в делокализцию заряда суммируются. Поэтому, например, исключительно высокой стабильностью обладают триарилметильные ионы. Так, и борфторид трифенилметил-катиона (35), и трифенилметилнатрий (36) имеют чисто ионную структуру — случай, достаточно редкий для соединений, несущих заряд на атомах углерода.
Исключительно эффективная стабилизация при сохранении высокой реакционной способности достигается в биполярных ионах, в которых атомы или группы, несущие противоположный заряд, непосредственно соседствуют в структуре. Таковы илиды — биполярные ионы, в которых карбанион-ный центр стабилизирован соседним положительно заряженным ониевым центром на атомах фосфора, серы, азота, мышьяка. Примерами типичных илидов могут служить соединения типа фосфонийметилида (37) или сульфо-нийметилида (38).

В последние десятилетия в обиход органического синтеза уверенно входят многочисленные ионные реагенты, в которых стабилизация заряда достигается за счег совершенно иных эффектов, чем те, что рассматривались выше в терминах классической органической химии. В этих реагентах основным фактором, определяющим стабилизацию заряда на атоме углерода, является наличие фрагментов, содержащих комплексы переходных металлов [5]. Некоторые примеры реагентов этого типа будут рассмотрены в последующих разделах.
Две другие возможности стабилизации органических ионов связаны с факторами внешней среды, в которых находятся активные частицы. Важнейшим из таких факторов являтся природа противоиона. Если противоион обладает низкой реакционной способностью и (что часто взаимосвязано) высокой дс-локализацией заряда, то это затрудняет его ассоциацию с органическим ионом с образованием ковалентной связи и повышает стабильность такой ионной системы. В этом смысле хорошими противоионами для карбокатионов являются анионы типа перхлората (39) или трифлата (40), а также такие координационно насыщенные анионы, как тетрафторборат (41), гексахлорантимонат (42) или гексафторфосфат (43). Наиболее обычными противоионами для карбанионов являются катионы щелочных металлов, особенно связанные в комплексы с краун-эфирами (см. выше), а также стабильные органические катионы типа координационно насыщенного тетрабутиламмоний-катиона (44).
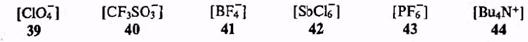
Природа растворителя является еще одним важнейшим внешним фактором, влияющим на стабильность органических ионов. Многостороннее по своему характеру влияние растворителя в первом приближении можно схематически свести к двум аспектам. С одной стороны, полярный растворитель, т.е. жидкость с высокой диэлектрической проницаемостью, чисто физически снижает кулоновское взаимодействие зарядов. Этот эффект может быть довольно значительным: например, переход от неполярного растворителя (гексана) к полярному (ацетонигрилу) уменьшает силы кулонов-ского взаимодействия в 21 раз. С другой стороны, молекула растворителя может стабилизировать ионы любого заряда за счет заряд-дипольных взаимодействий, образования водородных связей, комплексов разного типа, короче, за счет эффектов, обобщенно обозначаемых термином сольватация. Эти эффекты сольватации приводят к значительному экранированию заряда иона молекулами растворителя и частичной делокализации заряда, распределению его между ионом и сольватной оболочкой.
Любой способ стабилизации ионов путем делокализации заряда, будь то за счет групп, входящих в состав иона, или за счет сольватации, разумеется, тем эффективнее, чем значительнее смещение электронов к катионнму центру карбокатионов или от анионного центра карбанионов. Такая делокали-зация не должна, однако, переходить некоторые пределы, за которыми может произойти разрыв старых и/или образование новых ковалентных связей. Один подобный пример мы уже приводили: в mpe/я-бутильном катионе (26), Стабилизация которого осуществляется за счет поляризации связей С-Н, при неблагоприятных условиях может произойти полный сдвиг заряда, результатом чего будет выброс протона и образование двойной связи. Аналогичные примеры можно найти и в химии карбанионов. Так, например, известно, что при наличии хлора как заместителя при карбанионном центре стабильность этого аниона существенно возрастает из-за высокой электроотрицательности хлора. По этой причине трихлорметилъный анион (45), легко генерируемый при действии оснований на хлороформ, относится к категории стабильных анионных частиц, и известно немало реакций, протекающих с участием этого интермедиата. Однако эффект стабилизации для этой частицы за счет сдвига электронов на атомы хлора, очевидно, несколько «зашкален», поскольку для 45 особенно характерна склонность к элиминированию хлор-аниона и образованию дихлоркарбена (46).
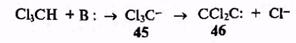
Типичные карбокатионы и карбанионы даже при значительной делока-лизаиии заряда все еще сохраняют чрезвычайно высокую реакционную способность. Это обстоятельство приводит к двум очень важным с точки зрения органического синтеза следствиям.
1. Карбокатионы способны реагировать не только с анионами, но и с ко-валентными поляризованными молекулами, атакуя при этом отрицательный конец диполя. Аналогично карбанионы охотно атакуют положительный конец диполя поляризованных ковалентных молекул. Так, типичный карбани-он 47 легко реагирует с ковалентной молекулой алкилгалогенида, атакуя, естественно, атом углерода как положительный конец диполя этой молекулы, что и приводит к образованию новой связи С—С.
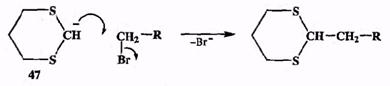 Схема 2.16
Схема 2.16
|
2. Многие органические содинения, являясь ковалентными в самом строгом смысле этого понятия, тем не менее благодаря очень значительной поляризации одной из связей могут в некоторых реакциях вести себя наподобие оргнических ионных соединений. Например, ковалентные, но сильнополя-ризованные соединения, такие, как метилтрифлат (48) или метилитий (49), в большинстве реакций ведут себя так, как если бы в их составе находились метил-катион или метил-анион соответственно.

Практически эти два обстоятельства означают следующее. Прежде всего, очевидно, что в гетеролитических реакциях образования связи С-С, которые описываются формальной схемой ассоциации карбокатиона с карбанионам, вовсе не обязательно, чтобы оба компонента были действительно ионными. Вполне достаточно, если лишь один из этих компонентов представлен стабилизированным ионом, а второй может быть просто поляризованной ковалентной молекулой с противоположным по знаку частичным зарядом на атоме углерода. Более того, во многих реакциях оба компонента могут быть ковалентными, а не ионными соединениями, если поляризация связи в одном из них достаточно велика или может стать таковой под действием соответствующих катализаторов или реагентов. В любом из этих случаев конечный результат реакции з точности соответствует той идеализированной схеме ретросин-тетического анализа, с которой мы начинали этот раздел и которая предусматривала разрыв связи С—С с образованием карбокатиона и карбаниона.
Идя по пути постепенного «развенчания» роли карбокатионов и карбани-онов в органическом синтезе, следует сделать еще один шаг. Некоторые ковалентные соединения даже со слабополяризовнными связями могут выполнять роль ионных реагентов, если их электронные системы легко поляризуемы. В таких молекулах приближение ионного или даже просто сильнополя-ризованного реагента может вызывать столь значительное смещение электронов, что в момент реакционного акта может образоваться интермедиат (и/или переходное состояние) ионного типа, подобно тем, которые могли бы образоваться в реакциях ионных реагентов. Типичные примеры подобного рода реакций — уже обсуждавшиеся выше случаи электрофильного замещения в толуоле (см. схему 2.3). В этих реакциях электронейтральная и лишь слабополяризованная молекула, толуол, выступает в роли эффективного эквивалента соответствующего карбаниона MeС6Н4-.
– Конец работы –
Эта тема принадлежит разделу:
ОРГАНИЧЕСКИЙ СИНТЕЗ
ORGANIC SYNTHESIS... THE SCIENCE BEHIND THE ART...
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Органические ионы и факторы, определяющие их стабильность
Что будем делать с полученным материалом:
Если этот материал оказался полезным ля Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.027 сек.
Новости и инфо для студентов