Реакция Вюрца. Аллильное сочетание и родственные случаи - раздел Образование, ОРГАНИЧЕСКИЙ СИНТЕЗ Выше Мы Уже Обсуждали Реакцию Вюрца Как Один Из Простейших Случаев Образовани...
Выше мы уже обсуждали реакцию Вюрца как один из простейших случаев образования связи С—С. В этой реакции одна молекула алкилгалогенида выступает в роли элекгрофила (эквивалента карбокатиона), в то время как вторая под действием металла превращается в соответствующий алкилметал, который исполняет роль нуклеофильной компоненты сочетания (эквивалента карбаниона). Отмечалось также, что эта давно известная реакция была модифицирована (за счет изменения природы нуклеофильной компоненты, т.е. перехода к использованию купратных реагентов) таким образом, что в настоящее время сочетание по схеме реакции Вюрца может считаться действительно общим методом синтеза.
С учетом сказанного легко спланировать модельный синтез, скажем я-бу-тана, соответствующий какой-либо из альтернативных схем разборки по ге-теролитическому механизму:
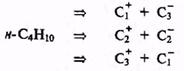
Все эти три схемы в общем равноценны и вполне реальны, так как требуемые электрофилы, например СН3I, С2Н5I и С3Н7I, и нуклеофилы, например CH3MgI, C2H5MgI и C3H7MgI (модифицированные солями меди), легко доступны. Можно, конечно, возразить, что синтезировать н-бутан вообще не нужно, так как этот углеводород в изобилии поставляется нефте- и газоперерабатывающей промышленностью. Тем не менее, эта задача может реально возникнуть, если для исследовательских целей неободимо иметь н-бутан, содержащий метку (например, дейтерий или 13С) в заданном положении. В этом случае выбор подходящей пары электрофил+нуклеофил будет определяться более всего доступностью соответствующих меченых предшественников.
Однако в синтезе чаще встречаются задачи, в которых формально возможные альтернативы на самом деле не являются равноценными. Рассмотрим, например, синтез углеводорода 50 (схема 2.17). В этой молекуле имеют
 Схема 2.17
Схема 2.17
|
ся два легко распознаваемых структурных элемента, которые входят в состав многих легкодоступных соединений, а именно: бензольное ядро и остаток метилацетилена. Очевидно, что в синтезе 50 разумно использовать исходные вещества, уже содержащие эти фрагменты. Учет таких соображений позволяет предложить два варианта разборки целевой молекулы, показанных на схеме 2.17 (путь «о» и путь «*»).
Разборка по связи бензил — этинил (путь «а») приведет нас к двум парам ионов 51 + 52 или 53 + 54. Для бензил-аниона (51) легко найти синтетический эквивалент в виде бензилмагнийбромида. Однако не имеется очевидного реагента, способного служить эквивалентом этинильного катиона 52, и поэтому можно исключить из дальнейшего рассмотрения вариант *at» как мало перспективный. Напротив, вариант «а2» вполне реален, так как для обоих ионов 53 и 54 имеются хорошо известные эквиваленты в виде бензил-хлорида и натриевого производного метилацетилена соответственно. Примерно такая же ситуация возникает при анализе разборки по связи фенил — пропаргил (путь «6»). Здесь также можно рассматривать варианты сборки этой связи из пар ионов 55 + 56 или 57 + 58.
Для реализации первого из этих вариантов «Z>i» необходимо подобрать синтетический эквивалент для фенил-катиона (55), что отнюдь не просто (для аниона 56 таким эквивалентом служит соответствующий пропаргиллитиевый реагент). Напротив, в варианте «Ьг» нуклеофильный 57 и элсктрофильный 58 компоненты легко идентифицируются в виде соответственно фенилмагний-бромида (59) и пропаргилхлорида (60). Таким образом из четырех формально равноценных вариантов решения задачи синтеза 50 мы приходим к двум — варианты *а2» и «fa», и оба они реально осуществимы (см. схему 2.17).
В реакциях, которые мы только что разбирали, фигурируют очень типичные синтетические эквиваленты органических ионов — это нутслеофилы (реагенты Гринъяра и ацетилениды) и электрофилы (бензил- и пропаргилгало-гениды). Общей чертой этих электрофилов является наличие системы тг-электронов по соседству с потенциальным карбокатионным центром. Это обеспечивает повышенную легкость замещения по этому центру и, поэтому реагенты, содержащие бензильную (61), пропаргильную (63) или аллильную (62) группировки, особенно эффективны как эквиваленты карбокатионов.
Учет этого обстоятельства указывает на то, что в общем случае ретросинтети-ческий анализ целевых структур, содержащих кратные углерод-углеродные связи или ароматические ядра, целесообразно начинать с разборки связи С-С у ал-лильного, пропаргильного или бензильного центров с тем, чтобы выйти к паре бснзил(аллил- или пропаргил-)-катион + карбанион и уже далее анализировать доступность реагентов, необходимых для реализации такой схемы [6].
Проиллюстрируем сказанное на конкретном примере синтеза непредельного спирта 64 — полового аттрактанта яблоневой плодожорки Laspeyresia pomonella, распространенного вредителя яблоневых садов [7] (схема 2.18). Разборка структуры 64 по связи у аллильного атома углерода привела к ал-лильному катиону 65 и функционализованному алкильному аниону 66. Очевидными эквивалентами этих ионов служили бромид 61 и реагент Гриньяра
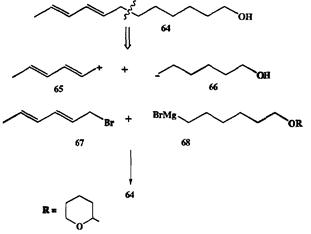 Схема 2.18
Схема 2.18
|
68, содержащий защищенную (в виде дигидропиранильного производного) гидроксильную группу. И электрофил 67, и нуклеофил 68 легко получались из доступных исходных веществ, и их конденсация протекала с хорошим выходом. После этого потребовалось лишь снять защиту, чтобы получить целевой продукт 64.
Подвижная система л>электронов кратных связей С—С, ответственная за стабильность соответствующих карбокатионов, оказывает столь же эффективное действие на стабильность карбанионов. Вследствие этого разборка по аллильной, бензильной или пропаргильным связям выигрышна еще и потому, что получающийся при этом непредельный фрагмент может быть представлен не только как карбокатион, но и как карбанион. Подобный дуализм всегда полезен, поскольку он расширяет область поиска наиболее подходящих вариантов, но особенно часто им пользуются при синтезе целого рада представителей одного из важнейших классов природных соединений, а именно ациклических изопреноидов*.
Структуры многих представители этого класса соединений выглядят так, как будто они были специально предназначены для ретросинтетического анализа с разрывом связи С-С между аллильными атомами углерода.
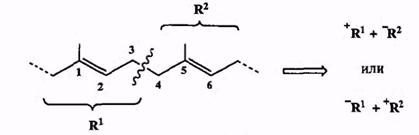 Схема 2.19
Схема 2.19
|
В самом деле, типичные представители этих природных соединений содержат 1,5-диеновую систему, разборка которой по центральной связи С-3—С-4 автоматически приводит к двум аллильным фрагментам (схема 2.19). Любой из них может в принципе рассматриваться как карбокатион или карбанион. Образование связи С-С путем сочетания двух таких фрагментов может определенно считаться «верным делом», и по этой причине во множестве синтезов изопреноидов используются в качестве «строительных блоков» аллильные заготовки самого причудливого строения. Выбор конкретной природы реагентов на этой ключевой стадии определяется, в основном, доступностью соответствующих исходных соединений. При этом, правда, приходится еще считаться с возможностью аллильной перегруппировки как в карбокатионах, так и карбанионах, что накладывает некоторые ограничения и на природу используемых реагентов, и на условия проведения такого сочетания**.
Все темы данного раздела:
ОРГАНИЧЕСКИЙ СИНТЕЗ
НАУКА И ИСКУССТВО
Перевод с английского
профессора, д-ра хим. наук В. А. Смита
и профессора, д-ра хим. наук А. Ф. Бочкова
Редакция литературы по химии
ISBN5-03-003380-7 (русск.)
ISBN 0-85404544-9 (англ.)
© The Royal Society of Chemistty 1998
© Перевод на русский язык, оформление «Мир», 2001
© OCR сканированной кн
Цель однозначна и бесспорна
С древних времен человеку были известны чарующие цвета, которые придавали тканям природные красители, добываемые из различных растений и животных. Уже в XIII в. до н. э. финикийцы владели искусств
Цель однозначна, но не бесспорна
Однако важность того или иного направления в науке чаще всего не может быть оценена столь прямолинейно только по критерию немедленной полезности конкретных научных исследований. На протяжении всей
Синтез как поиск (цель бесспорна, но не однозначна).
Синтез природных веществ, в том числе обладающих полезными свойствами, — это лишь одна, наиболее очевидная, но далеко не единственная задача органического синтеза. Как показывает в
Синтез как инструмент исследования
Во всех обсуждавшихся выше примерах синтез выполняет чисто препаративную функцию, т.е. поставляет нужные вещества. В принципе для решения таких задач не имеет значения, каким именно путем было по
Строение соединений с их свойствами
Пожалуй, главная, наиболее фундаментальная задача не только органической химии, но и всей химической науки — это установление зависимости свойств вещества (физических, химических, биологических) ка
Создание новых структур, проблемных для органической химии
На протяжении всей истории органической химии в ней возникали и продолжают возникать проблемы теоретического характера, для решения которых необходимо было изучить те или иные соединения с экзоти
Расширение круга известных органических соединений
Это — одна из традиционных и наиболее скромных сторон деятельности химиков-синтетиков. Скромных потому, что большинство таких синтезов носит весьма заурядный характер, и уже давно никого не удивля
Вводные замечания
Название этой главы может создать впечатление, что мы собираемся рассмотреть в ней все или хотя бы большинство методов, используемых в современной синтетической практике. Надо сра
Возможность протекания органической реакции. Общие соображения
Во введении мы упоминали привлекательный, но абсолютно нереальный путь синтез уксусной кислоты из метана и углекислого газа (диоксида углерода):
СН4 + СО2 → СН
Термодинамическая допустимость реакции
Ископаемое сырье, служащее в конечном счете основным исходным материалом для органического синтеза, образовалось в результате чрезвычайно длительных биогеохимических процессов. За это время оно ус
Термодинамический и кинетический контроль
Для того чтобы термодинамически допустимое превращение X → Y могло осуществиться, реагирующая система X (это может быть одно вещество или несколько компонент, словом, все участники процесса),
Органическая реакция и синтетический метод
Термину «синтетический метод» трудно дать строгое определение, но не трудно описать смысл этого понятия. Идеальный синтетический метод может быть уподоблен оператору в математике,
Принципы сборки связи С-С. Гетеролитические реакции
Основу типичной органической молекулы, ее углеродный скелет, составляет система непосредственно связанных друг с другом атомов углерода. Поэтому методы создания углерод-углеродных с
Органические ионы и факторы, определяющие их стабильность
Высокая химическая активность карбокатионов и карбанионов связана прежде всего с силами кулоновского взаимодействия. Точечный заряд, сосредоточенный на атоме углерода, создает элек
Электрофилы и нуклеофилы в реакциях образования связей С-С
Существование обширных классов органических реакций, которые могутбыть формально описаны в терминах ионных схем, но в которых реально участвуют ковалентные соединения, позволяет говорить об экви
Карбонильные соединения как нуклеофилы и электрофилы
В определенном смысле карбонилсодержащий фрагмент С-С=О может рассматриваться как аналог аллилъной системы С—С=С. Однако в отличие от последней в карбонильных соединениях эффективная стабилизация
Карбометаллирование алкинов
Изложенные выше принципы проведения нуклеофильного присоединения по кратным связям как последовательности независимых стадий атаки нуклеофила и взаимодействия образующегося карбанионного интермеди
Ретросинтетический анализ ациклических целевых структур. Общие рекомендации.
Выше мы рассмотрели лишь некоторые наиболее типичные и часто употребляемые методы сборки связей С-С и С=С. Эта выборка, несмотря на ее Неизбежную ограниченность, дает возможность сформулировать ря
Карбокатионные или карбанионные реагенты. О некоторых дополнительных возможностях проведения реакций образования связи С-С
Вначале разд. 2.2.3 мы не делали никаких принципиальных различий между карбокатионами и карбанионами, рассматривая и те, и другие в качестве равноправных партнеров в гетеролитичес
Взаимопревращения функциональных групп
До сих пор мы рассматривали лишь те реакции, результатом которых является образование новой связи С—С, и почти ничего не говорили о возможности переходов от одного типа органически
Изогипсические трансформации. Синтетическая эквивалентность функциональных групп одного уровня окисления.
Как мы уже могли убедиться, функциями, наиболее часто возникающими при сборке связи С—С, являются спиртовая (реакции Гриньяра, альдольная конденсация) и олефиновая (реакция Виттига, кротоновая конд
Неизогипсические трансформации как пути переходов между различными уровнями окисления
В этой группе наиболее значимыми для синтеза являются такие превращения кислородсодержащих соединений, как окисление спиртов до карбонильных соединений или карбоновых кислот и обратные им превращ
Взаимопревращение функциональных групп как стратегический метод в полном синтезе.
В начальный период развития органического синтеза было естественно выстраивать синтетическую цепочку, используя в качестве исходного соединения то или иное вещество, выделяемое из
Селективность обеспечивается выбором подходящей реакции
Наиболее простой пример такого подхода мы рассматривали на примере бро-мирования толуола (см. разд. 2.1.3). Действительно, в толуоле имеются две функциональные группы, способные легко реагировать с
Варьирование природы реагентов как способ управления селективностью реакции
Хорошо известно, что даже в пределах одной и той же реакции относительная реакционная способность родственных функций может ощутимо зависеть от конкретных особенностей используемого реагента. Поэто
Альтернативных реакционных центров субстрата
Классический пример такого подхода к решению проблемы — ацетоуксус-ный эфир (168).Его обычной реакционноспособной формой является 1енолят 169,реакции которого с ра
Защита функциональных групп как универсальный способ управления селективностью реакций
Во всех подходах к проблеме селективности, которые мы рассматривали вы-ше, «игра» строилась на вариациях, непосредственно затрагивающих участ-ников основного процесса: изменялись природа субстрата
Идеальный органический синтез: фантастика или достижимая цель?
Пофантазируем немного на тему о том, каким бы хотелось видеть идеальный органический синтез (недалекого будущего?). Мы говорили о том, что синтез состоит в конструировании молекул.
Реагенты и синтетическая эквивалентность
Разумеется, аналогию между реагентом в синтезе и деталью какой-либо механической конструкции не следует понимать слишком буквально, хотя бы уже потому, что обычно реагент входитв собираемую структ
Понятие о синтонах
Обобщенное описание эквивалентности чрезвычайно полезно с сугубо прагматических позиций планирования органического синтеза, поскольку с его учетом резко расширяется поле выбора реагентов, применим
Синтонный подход как инструмент в разработке путей синтеза
Введение в обиход синтонов как элементарных блоков-заготовок предоставляет химику систему готовых решений если не всех, то многих тактических задач. Современный синтетик при анализе структуры цел
Изоструктурные синтоны обратной полярности
Как видно, синтонный подход позволяет планировать синтез на основе гете-ролитическях реакций как сборку целевой молекулы из готовых «кубиков», порядок сцепления которых определяется противоположнос
Специфика задач при синтезе циклических соединений
Вообще говоря, построение молекул, в состав которых входит замкнутая цепь углеродных атомов (цикл), требует решения уже знакомых нам задач образовния связей углерод-углерод. Почему
Малые циклы: производные циклопропана и циклобутана
В циклопропане валентные углы атомов, образующих цикл, равны 60', т. е. очень сильно отличаются от валентного угла тетраэдрического атома углерода (109,5°). Поэтому неудивительно, что энтальпия об
Пят- и шестичленные циклы
Благодаря минимальным искажениям валентных углов и минимальному напряжению, обусловленному взаимодействием несвязанных групп, пяти- и шестичленные циклы (как и ведущие к ним переходные состояния)
Циклы большего размера. Принципы макроциклизации. Эффекты многоцентровой координации
Число атомов в цикле (п)
Относительная скорость(при 50˚С)
1,5 10е
1,7
Циклоприсоединение - методы, специально созданные для получения циклических структур
Нетрудно заметить, что все ранее рассмотренные методы циклообразования имеют одну общую особенность: циклизация осуществляется как внутримолекулярная реакция замыкания единственной
Циклоприсоединение
Среди множества реакций, относящихся к этому классу, особое место занимает [4 + 2]-циклоприсоединение. Это — реакция Дильса—Альдера [2а], как правило, не требующая катализа или иницирования облуч
Циклоприсоединение в синтезе производных циклобутана
[2 + 2)-Циклоприсоединенис относится к категории важнейших синтетических методов, поскольку эта реакция позволяет получать различные производные циклобутана по схеме сборки из двух алкеновых фрагм
Синтез циклопропанов путем [2 + 1]-циклоприсоединения
Синтез трехчленных циклов по схеме циклоприсоединения должен, очевидно, включать взаимодействие непредельного субстрата, например алкена, с каким-либо Срреагентом, выступающим в роли синтетическог
Селективность циклообразования в комплексах переходных металлов
Вспомним, каким трудоемким путем (с общим выходом 0,75%) был впервые получен циклооктатетраен (137,схема 2.65). Этот 10-стадийный синтез был впоследствии воспроизведен другими иссл
Радикальные реакции и их роль в синтезе циклических соединений
Как мы уже отмечали, большинство методов образования связей С—С в полном синтезе основано на гетеролитических реакциях или на реакциях циклоприсоединения. Причины того, что гемолитические реакции
Расщепление связей С-С и перестройка углеродного скелета как синтетические методы
Выше мы обсудили основные типы реакций и методов, используемых для образования связей С-С углеродного скелета ацикличгских или циклических молекул. Этот набор должен быть дополнен еще группой мето
Расщепление одинарных связей С-С
Пожалуй, наиболее известный и очевидный пример конструктивной роли «деструктивной» реакции — декарбоксилирование алкилированных производных ацетоуксусного или малонового эфира. По
Синтетическое использование реакций расщепления двойной углерод-углеродной связи
Созидательный потенциал реакций, приводящей к разрыву углерод-углеродных связей, еще более наглядно может быть продемонстрирован на примере окислительного расщепления олефинов. Сре
Перегруппировки углеродного скелета и некоторые возможности их использования в полном синтезе
Конструктивные и деструктивные реакции, которые мы до сих пор рассматривали, отличаются тем общим свойством, что в них затрагиваются (разрываются или образуются) лишь связи тех атомов, которые не
Перегруппировка Кляйзена-Джонсона—Айрленда и гидрокси-перегруппировка Коупа
Как показано в общем виде на схеме 2.154, синтетический результат перегруппировки Кляйзена сводится к введению аллильного фрагмента по а-ато-му исходного карбонильного соединения через промежуточн
Трансформации малых циклов и их роль в полном синтезе
Как было показано выше, разработано множество методов, позволяющих получать циклы различных размеров, в том числе входящие в состав полициклического скелета. Размер цикла, который может быть обра
Заключительные замечания
В данной главе мы, конечно, не имели возможности сколько-нибудь полно обсудить все те методы, которые составляют основу тактики современного органического синтеза. Однако мы надеемся, что даже на о







Новости и инфо для студентов