рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Образование
- /
- ОРГАНИЧЕСКИЙ СИНТЕЗ
Реферат Курсовая Конспект
ОРГАНИЧЕСКИЙ СИНТЕЗ
ОРГАНИЧЕСКИЙ СИНТЕЗ - раздел Образование, ...

ORGANIC SYNTHESIS
THE SCIENCE BEHIND THE ART
W. A. Smit
Zelinsky Institute of Organic Chemistry, Moscow, Russia
A. F. Bochkov
Institute of Biochemical Physics, Moscow, Russia
R. Caple
University of Minnesota, Duluth, Minnesota, USA

| THE ROYAL SOCIETY OF CHEMISTRY Information services |
В. Смит, А. Бочков, Р. Кейпл
ОРГАНИЧЕСКИЙ СИНТЕЗ
Перевод с английского профессора, д-ра хим. наук В. А. СмитаББК 24.2

Издание осуществлено при финансов о и поддержке Российского фонда фундаментальных исследований по проекту № 00-03-46002
Издание выпущено в свет при участии Института органической химии
Им. Н.Д. Зелинского
Редакция литературы по химии
После выхода книги в свет вряд ли найдется более пристрастный читатель, чем сам автор (а здесь нас трое!). Пожалуй,…Цель однозначна и бесспорна
В Древнем Риме рецепт производства пурпура относился к категории наиболее тщательно охранявшихся государственных секретов. Согласно указу Нерона,… В 1878 г. Байер [1б] разработал недорогой метод синтеза, пригодный для… Эти поистине триумфальные достижения произвели сильнейшее впечатление не только на химиков, но и на общество в целом.…Цель однозначна, но не бесспорна
Конечно, справедливо утверждение, что во многих случаях сложность и нетривиальность структур природных соединений воспринимаются сами по себе как… Среди множества природных соединений существует обширный класс изопреноидов… Однако уже в 1960-х годах эти представления пришлось радикальным образом пересмотреть. В частности было установлено,…Синтез как поиск (цель бесспорна, но не однозначна).
Синтез природных веществ, в том числе обладающих полезными свойствами, — это лишь одна, наиболее очевидная, но далеко не единственная задача… Вероятно, исторически первой областью органической химии, которая быстро и… Следовательно, если задача будет сформулирована как синтез ярко-красного азокрасителя с основными свойствами, то…Синтез как инструмент исследования
Однако в некоторых областях синтез используется не просто в качестве удобного «подручного средства», а составляет самую суть задачи. Речь идет, в… Другой, классический подход к установлению структуры вещества основан на его… От всех упомянутых выше ограничений свободен встречный синтез. Если структура исследуемого вещества подтверждена его…Выяснение закономерностей, связывающих
Строение соединений с их свойствами
Создание новых структур, проблемных для органической химии
Отдельные случаи изомерии органических соединений были известны уже в начале 1830-х годов, но рациональная интерпретация этого феномена стало… Для проверки теории ароматичности в свое время было исключительно важным…Расширение круга известных органических соединений
Как и все фундаментальные науки, органическая химия исследует неизвестное. Поэтому не представляется возможным предсказать открытия в той или иной… Химики, получившие более 100 лет назад бензоат холестерина —типично рутинный… Типичным примером искусственного создания совершенно новой области для исследования может служить химия…Литература
1.(а) Более подробно о химической истории «королевского» пурпура см. обзор: McGovem P. E., MichelR. Н. Асе. of Chem. Res., 23,152 (1990);см. также: Hoffmann Я American Scientist, 78, 308 (1990); (b) Bayer A Ber., 11,2128 (1878).
2. Grebe C, Liebemm С Ber., 2, 332 (1869).
3. Корана X. Г., цитировано по лекции: «Полный синтез биологически функционального гена», в сб.: «Итоги и перспективы развития биоорганической химии и молекулярной биологии», Овчинников Ю. А., Колосов М. Н. (ред.). — М.: Наука, 1378, ее. 203 - 209.
A.Reichstein Т. A., GussnerA. Helv. Chim. Acta,18, 608 (1935).
5.(a) Bergstrom S. Science,157,382 (1967); (b) Обзоры в сб.: Curtis-Prior P. В. (ed.),
Prostaglandins: Biology and Chemistry of Prostaglandins and Related Eicosanoids,
Churchill Livingstone, Edinborough, 1988.
6. (а) Популярное изложение истории таксола см.: Bowman S. Chem. Eng. News, 1991, Sept.2, 11; см. также: Freemantle M., там же, 1994, Aug. I; (b) обзоры см.: Cuenard D., Gueritte- Voegelein K, Potter P. Ace. Chem. Res., 26,160 (1993); Nicolaou K. C, Guy R. K., Dai W. M. Angew. Chem., Int, Ed. Engl., 33,15 (1994); (c) Holton R. A., Kim H. В., Somoza C, Liang F, Biediger R. J., Boatman P. D. Shindo M,, Smith С. С, Kim S., Nadizpdeh H., Suzuki Y., Tao C, Vu, P., Tang S., Zhang P., Murthi К. K, Gentile L. N.. Uu J. H. I. Am. Chem. Soc, 116, 1599 (1994); (d) Nicolaou K. C, Claibome С F., Nantermet P. G, Couladorous E. A, Sorensen E. J. J. Am. Chem. Soc., 116, 1599 (1994); (e) см., например: BlechertS, Kleine-Klausing A. Angew, Cherr,., Int. Ed. Engl., 30,412(1991).
7. (а)О проблеме иммуномодуляторов см.: KesslerH., Mierke D. F., Donald D., FurberM. Angew. Chem., Int. Ed. Engl, 30, 954 (1991); (b) об истории проблемы см.: Stinson S. Chem. Eng. News, 1989, Feb. 6, 30; (c) Jones T. K, Mills S. G., Reamer R. A., Askin D., DesmondR., Ryan K. M., Volame R. P., Shinkai I. J. Am. Chem. Soc, 111, 1157 (1989); (d) Nakatsuka M., Ragar, J. A., Sammakia Т., Smith D. В., Uehling D. E., Schreiber S. L., J. Am. Chem. Soc, 112,5583 (1990); (e) Goulet M.T., Hedkey D. W. Tetrahedron Lett., 32, 4627 (1991); (f) Andrus M. В., Schreiber S. L. J. Am. Chem. Soc, 115,10420 (1993).
8. (а) Обзор см.: Siddal J. B. Chemical Aspects of Hormonal Interactions, Chemical Ecology, веб.: Chemical Ecology, Academic Press Inc., New York, 1970, Ch. 11, p. 281; (b) TrostB. M. Ace. Chem. Res., 3, 120 (1970).
9. (a) Gibberellins and Plant Growth, Krishnamurthy H. N. (ed.), Wiley, New York, 1975; (b) Corey E. J., DanheiserR. L., Chandrasekaran S., Keck G. E., Gopalan В., Larsen S. D., Siret, P., GrasJ. L J. Am. Chem. Soc, 100,8034 (1978).
10. Addicott E. Т., Lyon J. L, Ohkuma K., Thiessen W. E., Cams H. R., Smith O. £., Comforth J. W., MillborrowB. V., Ryback G, WareingP. F. Science, 159, 1493 (1968).
11. (a) ButenandtA., Hecker E., ffopp M., Koch W. Ann., 658, 39 (1962); (b) Truscheit £., EiterK. Ann., 658, 65 (1962).
12. Обзор см.: Kelly D. R., Chemistry in Britain, 1990,124.
13. (а) Выделение см.: Butler С. G., Callow R. K., Johnston S. С Nature, 184,1871 (1959); (b) синтез см.: Bellassoued M., Majidi A. Tetrahedron Lett., 32, 7253 (1991) и цитированные в этой статье работы.
14. Обзор см.: Pheromones, Birch M. С. (ed.), North-Holland Research Monographs, Frontiers in Biology, v.32, 1977, 595 pp.; см. также научно-популярные статьи: «Slave-making ants», Topqff H. American scientist, 78, 520 (1990); «Empire of the ants», Wilson E. D. Discoverer, № 3, 44 (1990).
15. (а) Об истории открытия и механизме действия, см.: Jaenicke L, Boland W. Angew. Chem., Int.Ed. Engl., 21,643 (1982); (b) синтез: Abraham W. D., Cohen T. J. Am. Chem. Soc., 113, 2313 (1991); Сгоше G. D., PaguetteL. A. J. Org-Chem.,46,4272(1981) и цитированные л этой статье работы.
16. Синтез см.: Corey Е. /., Achiwa К., Katzenellenbogen J. A. J. Am. Chem. Soc,, 91, 4318 (1969).
17. MacMorris Т. С, Barksdak A. W. Nature, 215, 320 (1967); Arsenault G. P., Biemann K., Barksdak A.W., MacMorris Т. С J. Am. Chem. Soc., 90, 5635 (1968).
18. (а) Выделение см.: Cook С. E., Whichard L. P., Turner В., Wall M. E., Egley E. H. Science, 154, 1189 (1966); (b) синтез см.: Johnson A. W., Govda G., Hassanali A., Knox J., Mcnako S., Razavi Z., Rosebery G. J. Chem. Soc, Perkin Trans I, 1981, 1734; (c) ChangM., Netzly D. If., Butler L. G., Lynn D. G. J. Am. Chem. Soc, 108, 7858 (1986).
19. Подробнее об этом см.: Барбье М. Введение в химическую экологию. — М.: Мир, 1978, 229 с; см. также: Levinson G. Naturwissenshaften, 59,477 (1972); Gerout V., в сб.: Progress in Photochemistry, v.2, J. Wiley & Sons, London, 1970, p. 143.
20. Обзор по ангифидантам и возможностям их использования в защите растений см.: LeyS. V., TcogoodP. L. Chemistry in Britain, 1990, January, 31.
21. Eisner Т., MtinwaldJ. в сб.: Pheromone Biochemistry, Prestwich G. D., BloomquistG. J. (eds.), Academic Press, Orlando, 1987, ch. 8, p. 251; очень живое наложение истории этих работ можно найти в популярной статье: Meinvrald J. Engineering & Science, 49, №5, 14(1986).
22. (a) CoreyE. J., DittamU. P. J. Am. Chem. Soc, 107, 256 (1985); (b) Corey E. J., Guzman-PerezA., NoeM. С J. Am. Chem. Soc., 116, 12109 (1994).
23. Об истории открытия см.: Roberts R. M. Serendipity, J. Wiley, New York, 1989, ch. 24, p. 159.
24. (a) Sarrett, L. H. J. Biol. Chem., 162, 591(1946); (b) краткая история работ по созданию стероидных препаратов изложена в обзоре: Hirschman R. Angew. Chem., Int. Ed. EngL, 30, 1278 (1991); (с) обзор см.: Шоппи К. У. «Стероиды», в сб.: Перспективы развития органической химии. — М.: ИЛ, 1959, с, 223.
25. (а) Обзор см.: УокерД. «Химиотерапия», ссылка [24с], с. 301; (Ъ) Klayrran D. L. Science, 228, 1049 (1985); (с) см., например: Avery M. A., Chong W. К. М., lennings-White С. J. Am. Chem. Soc, 114, 974 (1992); см. также: Ye В., Wu Y.-L J. Chem. Soc, Chem. Comm., 1990, 726; (d) Posner G. N.. Oh С H,, Milhous W. K. Tetrahedron Lett., 32, 4235 (1991) и цитированные в этой статье работы.
26. (a) Still W. С. J. Am. Chem. Soc, 101, 2493 (1979); (b) Hauptman H., Muhlbauer G., Sass H. Tetrahedron Lett., 27, 6189 (1986); (c) Mori M., Okada K., Shimezaki K., Chwnan T. Tetrahedron Lett., 31, 4037 (1990).
27. (а) Популярное изложение истории открытия и исследования см.: Stinson S. Chem. Eng. News, 1995, Apr. 17, 22; (b) Barret/A. G. M., Kasdorf K., Tustin G. J., Williams D. J. J. Chem. Soc, Chem. Comm., 1995, 1143; (c) Kuo M. S., Zielinsky R. J., Cialdella J. I., Marschke С. К., Dupuis M. J., Li G. P., Kloosterman D. A., Spilman С. Н., Marshall V. P.i. Am. Chem. Soc, 117, 10629 (1995).
28. (а) Основные ссылки и общий обзор см.: Cross P. E., Dickinson R. P. Chemistry in Britain, 1991, 911; (b) BhagwatS. S., Hamann P. R., Still W. C, BuntingS., FitzpatrickF. A. Nature, 315, 511 (1985); (c) Bundy G. L, Tetrahedron Lett., 1975, 1957; см также: Nicolaou К. С, Magolda Я L, Smith J. B,, Aharony D., Smith E. F., LeferA. M. Proc Natl. Acad. Sci. USA, 76, 2566 (1979).
29. Bydeopd Р.Б. «Синтез», ссылка [24c], c.l 19.
30. Corey E. J., Angew. Chem., Int. Ed. Engl., 30, 455 (1991).
31.(a) Gomberg M., J. Am. Chem. Soc, 22, 757 (1900); (b) обзор см.: Gomberg M Chem. Rev., 1, 91 (1925); (c) Lankamp H., Nauta W. Т., MacLean С Tetrahedron Lett., 1968, 249: (d) изложение поучительной истории загадки гексафенилэтана можно найти в обзоре: McBride J. M. Tetrahedron, 30, 2009 (1974).
32. (a) Sachse H. Ber., 23, 1363 (1890); (b) Barton D. И. R. Experientia, 6, 315 (1950); (с) Бортом Д. Р. «Стереохимия», ссылка [24с], с. 57.
33. Цитировано по ссылке [29], с. 133.
34. История открытия тефлона увлекательно изложена в книге: Roberts R. M. Serendipity. Accidental Discovery in Science, J. Wiley & Sons, New York, 1989, p. 187.
35. Характерные примеры можно найти в обзорах: Mann J. Chem. Soc. Rev 16 381 (1987); Welch J. Т. Tetrahedron, 43, 3123 (1987).
36. Краткое изложение истории основных направлений в изучении проводящих полимеров см.: Kanatzidis M. G. Chem. Eng. News, 68, № 49, 36 (1990).
37. Обзор см.: Williams J. M., Beno M. A., Wang H. П., Leung P. С. W., Emge T. J., Geiser U., Carlson К. D. Асе. Chem. Res., 18, 261 (1985).
38. Hoffmann R. The Same and Not the Same, Columbia University Press, New York, 1995 (имеется перевод: Хоффман Р. Такой одинаковый и разный мир. — М.: Мир, 2001).
39. Seebach D. Angew. Chem., Int. Ed. Engl., 29, 1320 (1990).
Глава 2
Тактика синтеза
Вводные замечания
Название этой главы может создать впечатление, что мы собираемся рассмотреть в ней все или хотя бы большинство методов, используемых в современной… Глава начинается с изложения некоторых базовых представлений о химических… Для большинства синтетических задач ключевой проблемой является образование связи углерод — углерод, и поэтому мы…Каким образом может быть достигнуто требуемое превращение
Возможность протекания органической реакции. Общие соображения
СН4 + СО2 → СН3СООН А почему, собственно говоря, этот путь нереален? Казалось бы, и элементный… Первая причина (термодинамический запрет) подобна той, по которой санки легко скатываются с горы, но никогда…Термодинамическая допустимость реакции
Для того чтобы создать такие неравновесные системы, необходимо произвести некоторую работу, энергия для которой должна быть привнесена извне. Это… Источником химической энергии служат химические реагенты, энергия которых была… С термодинамической точки зрения органический синтез может быть уподоблен сложному и опасному путешествию в горах со…Наличие канала реакции.
Термодинамический и кинетический контроль
-> Вообще говоря, барьер между исходными веществами и продуктами реакции… Рассмотрим ту же систему X →Y, прибавив еще один компонент — термодинамически более стабильный продукт Z (рис.…Органическая реакция и синтетический метод
Термину «синтетический метод» трудно дать строгое определение, но не трудно описать смысл этого понятия. Идеальный синтетический метод может быть… Ключевая реакция метода может предполагать использование нестабильных… 1. RHal + Mg → R-MgHalОбразование связи С-С: ключевая тактическая проблема органического синтеза
Принципы сборки связи С-С. Гетеролитические реакции
Основу типичной органической молекулы, ее углеродный скелет, составляет система непосредственно связанных друг с другом атомов углерода. Поэтому… Для простой связи С-С возможна разборка на два противоположно заряженных иона… Оба эти типа частиц содержат большой запас энергии, и поэтому такие процессы в общем случае должны быть быстрыми,…Органические ионы и факторы, определяющие их стабильность
Высокая химическая активность карбокатионов и карбанионов связана прежде всего с силами кулоновского взаимодействия. Точечный заряд,… Далее, воздействие заряда иона на неионные молекулы усиливает поляризацию их связей и создает предпосылки для полной…Электрофилы и нуклеофилы в реакциях образования связей С-С
Электрофилы (Е) — реагенты, способные принимать пару электронов, а нуклеофилы (Nu) — реагенты, способные отдавать пару электронов с образованием… Однако для того, чтобы соединение вело себя как электрофил, не обязательно… Реакционная способность электрофильных и нуклеофильных реагентов может изменяться в очень широких пределах, и их…Реакция Вюрца. Аллильное сочетание и родственные случаи
С учетом сказанного легко спланировать модельный синтез, скажем я-бу-тана, соответствующий какой-либо из альтернативных схем разборки по… Все эти три схемы в общем равноценны и вполне реальны, так как требуемые электрофилы, например СН3I, С2Н5I и С3Н7I, и…Карбонильные соединения как нуклеофилы и электрофилы
Как мы уже говорили, в енолят-анионе заряд распределен между кислородом и а-углеродным атомом. Поэтому в общем случае его реакция с элект-рофилом… К счастью, направлением атаки электрофила можно управлять, варьируя природу реагента и/или условия проведения реакции.…Сопряженное присоединение к аф-непределъным карбонильным соединениям. Аннелирование по Робинсону и присоединение по Михаэлю с независимой вариацией аддендов
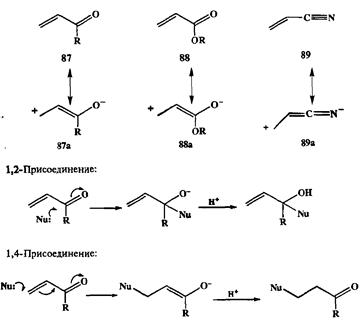 Схема 2.29
Схема 2.29
|
Рассмотренные выше реакции карбонильных соединений при всей широте их применения и синтетической значимости достигаемых с их помощью превращений далеко не исчерпывают тот огромный синтетический потенциал, который заложен в химии карбонильной группы. Качественно новые возможности появляются в системах, где карбонильная группа находится в сопряжении с двойной связью углерод—углерод. В таких структурах, типичными представителями которых являются соединения типа 87—89, я-электроны кратных связей образуют единую сопряженную систему, в пределах которой легко передаются эффекты поляризации связей. В силу этого подобные соединения в реакциях с нуклеофилами могут вести себя либо как уже знакомые нам карбонильные электрофилы, либо проявлять свойства электрофилов иного типа, в которых электрофильный центр локализован на р-углеродном атоме, как это отражено в канонических формулах 87а—89а.В результате подобные непредельные соединения, такие, как, например, <х,|3-непредельные кетоны или эфиры, могут подвергаться атаке нуклеофилом и по карбонильному атому углерода, и по р-углеродному атому (схема 2.29).
Первое из этих направлений нам уже хорошо известно и в случае показанной на схеме реакции с енонами его результатом является образование ал-лиловых спиртов — чрезвычайно полезных синтетических полупродуктов. Однако еще более интересные синтетические возможности открывает второй путь, 1,4-присоединение нуклеофилов, известное какреакция Михаэля [13]. В классическом варианте этой реакции в качестве таких нуклеофилов используют стабилизированные карбанионы, производные соединений типа малоново-го эфира или нитрометана, которые генерируют под действием слабых оснований непосредственно в реакционной среде в присутствии акцепторов Михаэля, например, метилвинилкетона (90), как это показано на следующих примерах:
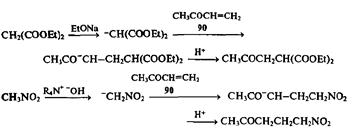
Реакция Михаэля представляет собой очень эффективный способ удлинения углеродной цепи электрофила на три (и более) атома углерода. Читатель, конечно, обратил внимание на то, что типичные акцепторы Михаэля, как, например, (90), — это продукты конденсации карбонильных соединений, которые могут быть получены по схеме альдольной конденсации (см. 73, схема 2.26), реакции Виттига (см. 82, схема 2.28), реакции Перкина или Манниха (см. ниже). Подчеркнем также, что типичными нуклеофильными компонентами реакции Михаэля служат ионные еноляты, производные карбонильных соединений. Таким образом, условия, требуемые для получения акцепторов Михаэля, очень схожи или даже идентичны условиям проведения самой реакции Михаэля. Эти обстоятельства создавали предпосылки для того, чтобы «состыковать» обе реакции — получение акцептора Михаэля и присоединение к нему нуклеофильного реагента — в связанную последовательность превращений, проводимых в одной колбе без выделения промежуточно образующихся продуктов. Более того, можно было ожидать, что функционально замещенные карбонильные соединения, типичные аддукты, получающиеся в результате реакции Михаэля, в тех же условиях могут быть далее вовлечены в такие типичные для них превращения, как, например, внутримолекулярная альдольная конденсация. Первым примером подобного согласованного проведения последовательности реакций карбонильных соединений явилосьаннелирование по Робинсону [14а,B], стандартный путь создания шестичленного цикла, — метод, широко применяемый в полном синтезе множества природных соединений. Типичный пример такой последовательности приведен на схеме 2.30.
Ключевая стадия показанной цепочки превращений — присоединение енолята 91 по двойной связи енона 90 [14с] (реакция Михаэля). Первичным продуктом этой реакции является тоже енолят-анион 92, способный к обратимой изомеризации в енолят 93. Нуклеофильный центр последнего пространственно сближен с имеющимся в молекуле электрофильным центром, карбонильной группой циклогексанового кольца, благодаря чему в условиях реакции достаточно легко протекает внутримолекулярная альдольная конденсация, сопровождающаяся дегидратацией, и в результате образуется би-циклический ендион 94. Показанный дикегон является одним из важнейших промежуточных полупродуктов в синтезе полициклических терпеноидов и стероидов, поскольку он содержит имеющуюся во многих из этих соединений систему А/В циклов, ангулярную метальную группу, кетонную группу в цикле В и еноновый фрагмент в цикле А, что обусловливает возможность реализации последующих синтетических трансформаций самых различных типов.
 Схема 2.30
Схема 2.30
|
Препаративные достоинства аннелирования по Робинсону очевидны. Последовательность стадий 90 + 91 -» 94 проводится в одну операцию. Более того, генерирование енолята из кетона 95 и акцептора Михаэля 90 из 96 также могут проводиться в одном реакционном сосуде. Последнее особенно важно из-за малой устойчивости метилвинилкетона (90) к длительному хранению. Генерация 90 in situ осуществляется посредством обработки четвертичной аммониевой соли 96 сильным основанием [14а]. Таким образом, если смесь реагентов 95 и 96 нагревать в присутствии сильного основания (амид натрия), то запускается показанная на схеме 2.30 последовательность превращений, даю-шая в конце концов бициклический дикетон 94. К сказанному можно добавить, что синтетический эквивалент енона 90, аммонийная соль 96, может быть в свою очередь легко получена из тривиальных соединений (ацетона, формальдегида и диэтиламина) по реакции Манниха [14d]) — реакции, в об щем-то очень близкой вышеописанным карбонильным конденсациям, с последующим N-алкилированием получаемого продукта метилиодидом.
Читатель мог заметить, что показанное на схеме 2.30 превращение моноциклического дикетона 95 в бициклический продукт 94 (циклогексаноанне-лирование) по сути дела представляет собой последовательность двух тривиальных реакций, а именно: присоединения по Михаэлю и альдольной конденсации. Почему же, в таком случае, этому синтетическому протоколу присвоено название «аннелирование по Робинсону»? Конечно, и альдольная конденсация, и реакция Михаэля были открыты задолго до исследований Робинсона. Однако безусловной заслугой последнего было осознание стратегических преимуществ синтетических решений, основанных на исчерпывающем использовании потенциала функций, имеющихся в исходных веществах. По сути дела Робинсоном была предложена новая стратегия построения шестичленного фрагмента с помощью тандемной последовательности простых и надежных реакций. Смысл этой стратегии по существу очень прост. Он заключается в том, что карбанионный интермедиат 92, образующийся на первой стадии реакции Михаэля, не перехватывается протоном из среды (как это обычно происходит в классической методике проведения реакции Михаэля) с образованием ковалентного продукта 92а, а используется на следующей стадии в качестве генерированного in situ нуклеофила, реагирующего (через стадию эквилибрирования с енолятом 93) с карбонильным электрофнлом по схеме внутримолекулярной альдольной конденсации (очевидно, что необходимым условием такого хода реакции является отсутствие в среде активных доноров протона). Это принципиальное преимущество можно в общем виде уяснить, если сравнить показанные на рис. 2.5 схемати ческие энергетические профили двух вариантов последовательностей для преврашения 95 в 94: (а) по схеме классической реакции Михаэля через ковалентный аддукт 92а и (B) по методу Робинсона через интермедиаты 92 и 93.
К настоящему времени разработано множество вариантов проведения тан-демных последовательностей превращений, аналогичных показанной на схеме 230, для самых разнообразных типов акцепторов Михаэля и а,р-непредельные карбонильных производных, и аннелирование по Робинсону считается сейчас одним из наиболее общих и надежных способов построения циклогексеноно-вого фрагмента почти в любом структурном контексте [14Ь,е]).
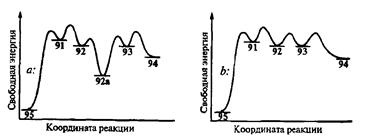 Рис 2.5
Схематические энергетические профили для двух вариантов образования 94 взаимодействием 95 и 90: классический, с раздельным выполнением стадий (а) и с выполнением обеих стадий в связной последовательности в одном реакционном сосуде (по Робинсону) (/>)
а — исходный дикетш 95 образует енолят 91, реакция которого с электрофилом 90 дает промежуточный карбанион 92. Последний присоединяет протон из среды и превращается в ко-валентное соединение 92а. Для выполнения следующей стадии необходимо снова затратить определенную энергию для генерации енолята 93, который и превращается далее в продукт 94.
B — исключается ненужный «спуск с горы» (92 -> 92а) и повторный подъем в гору (92а -» 93), а промежуточный енохят 92 непосредственно используется (через эквилибрирование с 93) на завершающей стадии.
Рис 2.5
Схематические энергетические профили для двух вариантов образования 94 взаимодействием 95 и 90: классический, с раздельным выполнением стадий (а) и с выполнением обеих стадий в связной последовательности в одном реакционном сосуде (по Робинсону) (/>)
а — исходный дикетш 95 образует енолят 91, реакция которого с электрофилом 90 дает промежуточный карбанион 92. Последний присоединяет протон из среды и превращается в ко-валентное соединение 92а. Для выполнения следующей стадии необходимо снова затратить определенную энергию для генерации енолята 93, который и превращается далее в продукт 94.
B — исключается ненужный «спуск с горы» (92 -> 92а) и повторный подъем в гору (92а -» 93), а промежуточный енохят 92 непосредственно используется (через эквилибрирование с 93) на завершающей стадии.
|
В рассмотренной связанной последовательности первая стадия является межмолекулярной реакцией акцептора Михаэля 90 с карбанионом, генерированном из кетона 91, а вторая — внутримолекулярной реакцией полученного енолятного интермедиата с электрофилом, карбонильной группой того же кетона 91. Уместно задаться вопросом, возможна ли реализация подобной же схемы раздельного присоединения к акцептору Михаэля для тех случаев, когда нуклеофил и электрофил не принадлежат одной и той же молекуле или, иными словами, когда обе стадии присоединения являются межмолекулярными? Ответ на этот вопрос нетрудно дать, если на основе приведенного выше рассмотрения аннелирования по Робинсону попытаться сформулировать в общем виде те условия, которые необходимы и достаточны для обеспечения такого хода реакции. Очевидно, что для этого, во-первых, необходимо проводить стадию присоединения нуклеофила по кратной связи акцептора Михаэля в отсутствие активных электрофилов (например, протона), способных немедленно «гасить» образующийся при этом карбанионный интермедиат. Это, в частности, означает, что реакцию надо проводить в апротонной среде. Во-вторых, необходимо также, чтобы этот интермедиат являлся стабильным, способным существовать в растворе как кинетически независимая частица вплоть до момента, когда в реакционную смесь будет прибавлен внешний электрофил. Естественно, требуется также ввести к минимуму возможность реакции образующегося нуклеофильного интермедиата с исходным электрофильным субстратом. Все эти условия могут быть соблюдены путем выбора соответствующих реагентов и условий проведения реакции, и во всех таких случаях реакция Михаэля может быть проведена как последовательность кинетически независимых стадий присоединения нуклеофила (Nu) и электрофила (Е) по связи С=С исходного субстрата или, иными словами, реализована в виде двух последовательных межмолекулярных реакций.
Сказанное выше является по сути дела всего лишь логическим следствием классической схемы описания процесса нуклеофильного присоединения по кратным связям как двустадийной реакции. Примечательно, однако, что на основе подобного анализа удалось разработать новую и очень продуктивную методологию, применимую для решения широкого круга синтетических задач. Одним из первых примеров успешного использования последовательности независимых стадии присоединения Nu и Е по двойной связи акцептора Михаэля был описанный Сторком [ 15а] синтез аддукта 97 (схема 2.31) — полупродуктав полном синтезе полициклического алкалоида ликоподина.
 Схема 2.31
Схема 2.31
|
Как показано на этой схеме, на первой стадии енон 98 реагирует с арил-магнийкупратным реагентом 99, давая продукт присоединения карбаниона Аг~, енолят 100.Последний при последующей обработке электрофилом, ал-лилбромвдом, превращается в конечный продукт 97. Суммарным итог показанного превращения — «сборка» целевой молекулы из трех предшественников с образованием двух новых связей С-С. Здесь также уместно подчеркнуть, что на примере этого синтеза бьшо показано принципиально важное значение применения купратных реагентов в качестве нуклеофилов на первой стадии реакции, присоединении по Михаэлю. Во-первых, реагенты этого типа (например, 99) обеспечивают практически полную селективность атаки по р-углеродноу атому сопряженной системы, что, как правило, трудно достижимо для реагентов типа литий- или магнийорганических соединений. Во-вторых, получающиеся в результате этой стадии купратные еноляты (например, 100)из-за своей пониженной основности и нуклео-фильности, являются существенно более стабильными интермедиатами по сравнению с соответствующими литий- или магнийпроизводными и менее склонны, чем последние, к участию в побочных реакциях. Благодаря этим особенностям именно купратные реагенты чаще всего используются в качестве нуклеофилов в реакциях типа присоединения по Михаэлю [ 15b-d].
К настоящему времени описаны многие десятки синтезов соединений различных типов, выполненных по обшей схеме трехкомпонентного сочетания, представленной выше для синтеза 97, с использованием самых разнообразных акцепторов Михаэля, нуклеофилов (как правило, купратного типа) и электро-филов. Благодаря показанной возможности почти неограниченных вариаций в природе всех участвующих компонент [15е], тандемная последовательность раздельных стадий — присоединения нуклеофила по Михаэлю/алкилирова-ние енолята — стала одним из самых эффективных стратегических приемов в современном органическом синтезе (подробнее об этом см. разд. 3.2.7).
Карбометаллирование алкинов
В своей основе этот метод представляет собой один из вариантов давно известной реакции нуклеофильного присоединения по тройной связи, которая… Так, например, присоединение литийкупратного реагента 101(схема 2.32) по…Ретросинтетический анализ ациклических целевых структур. Общие рекомендации.
1. Простая связь С—С в отсутствие близко расположенных функциональных групп. Схема 2.34 При этом эквивалентами карбанионов могут служить купратные комплексы литий-… 2. Простая связь С—С, один из атомов которой несет кислородный заместитель. Схема 2.35Карбокатионные или карбанионные реагенты. О некоторых дополнительных возможностях проведения реакций образования связи С-С
В силу сказанного становится понятным, почему большинство классических синтетических методов, описываемых в терминах ионных реакций, основаны по… Ранее мы уже отмечати, что классическая реакция Гриньяра может быть описана… Как аллилсиланы, так и силиловые эфиры енолов являются отличными нуклеофилами для реакций со стабилизированными…Взаимопревращения функциональных групп
До сих пор мы рассматривали лишь те реакции, результатом которых является образование новой связи С—С, и почти ничего не говорили о возможности… Реакции органических соединений, результатом которых являются трансформации… Начнем прежде всего с вопроса о том, что такое вообще «функциональная группа». В основе структуры типичных…Уровень окисления углеродного центра и
Классификация функциональных групп и их взаимопревращений
Можно использовать самые различные подходы для классификации функциональных групп и путей их взаимопревращений. Для наших целей наиболее подходящим кажется подход, основанный на рассмотрении состоянии окисления атомов, входящих в состав функциональных групп [19Ь]. Этот принцип и будет использован ниже, причем в первую очередь, естественно, нас будет интересовать состояние окисления атомов углерода в той или иной функциональной группе.
Как известно, окислением называют реакции, связанные с потерей атомом (или молекулой) электронов. Достаточно легко установить происходящие при этом изменения в состоянии окисления реагирующих партнеров для чисто ионных реакций. Однако для превращений ковалентных органических соединений понятия «окисление» или «восстановление» далеко не всегда кажутся столь же очевидными. Действительно, если речь идет об окислении первичного спирта в карбоновую кислоту (или обратном процессе), об окислении алкенов в эпоксиды или их превращении в алканы, то ясно, что это все — типичные окислительно-восстановительные реакции. Но уже классификация в тех же терминах таких реакций присоединения по двойной связи, как гидратация или бромирование, и обратных им реакций элиминирования не кажется столь же определенной. Тем не менее и по отношению к подобного рода реакциям можно уверенно использовать понятия окисления и восстановления, если опираться на определенные формальные критерии и принять за начало отсчета степень окисления углерода в алканах (уровень окисления 0).
Рассмотрим связь С-Н в алканах. Углерод более электроотрицательный элемент, чем водород. Вследствие этого электронная пара этой связи смещена к атому углрода, что в утрированной форме может быть представлено ионной формулой 122(схема 2.45). Нетрудно видеть, что при таком рассмотрении атом углерода в составе фрагмента С-Н аппроксимируется кар-банионом, которому таким образом и приписывается уровень окисления 0. К ионной системе такого типа уже однозначно применимы традиционные представления об окислительно-восстановительных реакциях. Так, окисление 122с переносом одного электрона приводит к радикалу 123,в го время как окисление с потерей двух электронов дает карбокатион 124.При такой трактовке переход от алканов к спиртам и далее к альдегидам и карбоновым кислотам может четко классифицироваться как процесс окисления с потерей двух, четырех или шести электронов и образованием функциональных производных уровней окисления I, 2 и 3 соответственно. Аналогичным образом можно интерпретировать переход от алканов к алкенам и алкинам (см. схему 2.45).
Такое рассмотрение создает вполне логичную основу для классификации функционально замещенных соединений как производных алканов, для чего требуется чисто формально рассмотреть изменение уровня окисления атомовуглерода последних, которое требуется для формирования той или иной функциональной группы или групп (см. схему 2.46).
 Схема 2.45
Схема 2.45
|
Еще более важным является то обстоятельство, что подобная классификация позволяет четко разделить все трансформации функциональных групп на два главных типа, а именно: изогипсические реакции, т. е. реакции, в результате которых не происходит изменений в уровнях окисления углеродных атомов, затрагивамых в этих превращениях, и неизогипсические реакции, результатом которых может быть окисление (повышение уровня окисления) или восстановление (понижение уровня окисления).
Оценку изменения уровня окисления органического соединения в ходе того или иного превращения проще всего сделать, если проследить за изменением уровня окисления соответствующего реагента. Так, например, образование спиртов в результате гвдратации алкенов, равно как и обратная реакция дегидратации безусловно относятся к категории изогипсических превращений, поскольку в них участвует вода, не играющая здесь роли окислителя или восстановителя. Напротив, любые варианты гидроксилиро-вания алкенов, ведущие к образованию 1,2-гликолей, описываются как формальное присоединение пероксида водорода, несомненного окислителя, и потому должны быть отнесены к разряду неизогипсических, окислительных реакций. Также неизогипсическими являются такие реакции, как присоединение водорода (восстановитель!) или брома (окислитель!) по двойным или тройным связям и соответствующие им обратные реакции (де-гвдриривание и дегалогенирование).
Следуя той же логике рассуждений, мы приходим к вывода, что образование литийорганичсских соединений или реактивов Гриньяра при действии металлов (восстановителей) на алкилгалогениды есть неизогипсическая восстановительная реакция, при которой субстраты с уровнем окисления 1 восстанавливаются до соединений уровня окисления 0, отвечающего алканам. Таким образом, получается, что металлоорганические соединения оказываются уникальными функциональными производными, имеющими нулевой уровень окисления. Этот довольно парадоксальный вывод подтверждается тем, что гидролиз этих соединений, т. е. заведомо изогипсическая реакция, приводит именно к углеводородам:
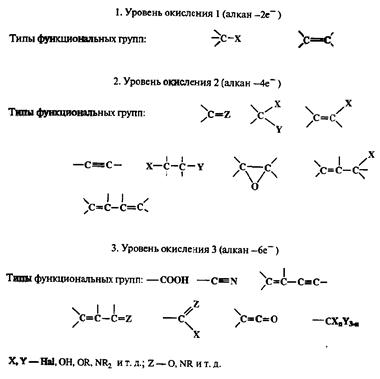 Схема 2.46
Схема 2.46
|
R-Hal + Mg → R-Mg-Hal
R-Mg-Hal + H2O → R-H + Mg(OH)Hal
Характеризуя в наиболее общем виде трансформационные переходы, отметим следующее:
1. Практически любые изогипсические трансформации легко осуществимы, так что взаимопревращения функциональных групп в пределах одного уровня окисления обычно не составляют проблемы.
2. Неизогипсичекие превращения осуществимы не для любых типов производных, а лишь для некоторых, особенно склонных претерпевать окисление или восстановление. Так. например, прямой переход от простых эфиров (уровень окисления 1) к ацеталям или кета-лям (уровень окисления 2) затруднен. Напротив, окисление спиртов в альдегиды или кетоны — это тривиальное превращение, равно как и обратный восстановительный переход. Приведем еще один пример: ацетилены легко превратить в олефины, в то время как аналогичный переход с уровня окисления 2 на уровень 1 для превращения дигалогеналканов в моногалогеналканы в общем случае затруднителен.
Таким образом напрашивается следующая аналогия: на каждом «этаже» (уровне окисления) можно свободно перемещаться из одной ячейки в другую. Напротив, перейти с «этажа» на «этаж» можно не в произвольном месте, а только предварительно добравшись до какого-либо «лифта», роль которого может выполнять далеко не каждая ячейка данного этажа. Такая картина при всей ее схематичности достаточно четко характеризует общие возможности и ограничения в использовании трансформаций функциональных групп. Это позволяет нам в дальнейшем изложении свести к минимуму рассмотрение конкретных реакций, обеспечивающих ту или иную трансформацию, и обратить внимание преимущественно на случаи, иллюстрирующие ограничения или, напротив, неочевидные возможности применения трансформаций функциональных групп в синтезе.
Изогипсические трансформации. Синтетическая эквивалентность функциональных групп одного уровня окисления.
С синтетической точки зрения важнейшими изогипсическими трансформациями спиртов являются образование алкилгалогенидов и сложных эфиров, включая… Особенно важными для синтетической практики являются такие производные… R-OSO2R1 + Nu- -> R-Nu + -OSO2R1Неизогипсические трансформации как пути переходов между различными уровнями окисления
Особо интенсивно разрабатывались новые методы в применении к задаче окисления спиртов. В цитированной выше монографии Ларока [19е] перечислено… Чаще всего окисление спиртов проводят с помощью хромового ангидрида и его… Диоксид марганца МпО2 является специфическим реагентом для окисления аллиловых спиртов до альдегидов (19Л. При этом…Взаимопревращение функциональных групп как стратегический метод в полном синтезе.
В начальный период развития органического синтеза было естественно выстраивать синтетическую цепочку, используя в качестве исходного соединения то… Но надо сказать, что подобного рода частичные синтезы, основанные на… Примером большей области, почти целиком построеной на частичных синтезах, может служить синтетическая химия углеводов.…Как управлять селективностью органических реакций
Классификация проблем селективности
Мы уже неоднократно говорили о селективности тех или иных органических реакций. Тем не менее, этот вопрос настолько важен для органического синтеза в целом, что он заслуживает более легального обсуждения как самостоятельная проблема.
Надежность синтетического метода не только предполагает, что данный метод может использоваться для эффективного осуществления требуемого превращения, но и подразумевает, что в избранных условиях между данной функциональной группой и реагентом протекает одна и только одна реакция. Тем не менее, этим проблема селективности далеко не исчерпывается. Дело в тем, что реальный субстрат может содержать несколько одинаковых или близких по свойствам функциональных групп, способных реагировать с одним и тем же реагентом, а по условиям задачи требуется провести превращение с одной из них, Кроме того, даже при наличии всего лишь одной функциональной группы, се превращение с использованием «чистой» (т.е. надежной) реакцииможет приводить к образованию нескольких изомерных продуктов. Характер проблем, связанных с селективностью, может быть весьма различен. Ниже мы рассмотрим некоторые типичные случаи, с которыми чаще всего приходится иметь дело в рамках решения задач обеспечения селективности тех или иных превращений. Если взглянуть на проблему селективности с точки зрения кинетики, то можно выделить три общих типа случаев, в каждом из которых возможно образование более, чем одного продукта в условиях данной реакции.
Тип 1. Последовательные реакции. На схеме 2.68 приведен ряд примеров таких последовательностей, в которых продукт, образующийся в результате первой реакции, способен подвергаться дальнейшему превращению в той же реакционной системе. Следовательно, в этом случае для достижения селективности требуемого превращения необходимо иметь возможность остановить процесс на какой-либо из стадий.
 Схема 2.68
Схема 2.68
|
Этого можно добиться разными способами. Например, обе реакции в превращении (1) принадлежат к одному и тому же типу гидрирования в присутствии гетерогенного катализатора. Поэтому для обеспечения селективного гидрирования ацетиленов в олефины необходимо модифицировать катализатор так, чтобы восстановление двойной связи на этом катализаторе проходило существенно медленнее, чем восстановление тройной. Этому требованию отвечает, например, катализатор Линдлара — палладий, осажденный на карбонате кальция и дезактивированный добавками оксида свинца (Pd-СаСОз—РЬО).
Напротив, стадии окисления первичных спиртов в альдегиды, а последних в кислоты [последовательноть (2)| резко различаются по своему механизму, что позволяет осуществить первую из этих реакций селективно за счет использования специфических реакций и реагентов. Для этой цели, например, очень эффективна система ДМСО — кислота Льюиса (см. схему 2.60), не способная окислять альдегиды.
Обеспечение селективности алкилирования енолятов [превращение (3)] — это одна из центральных проблем в синтетической химии карбонильных производных, для решения которой разработан комплекс разнообразных приемов, которые рассмотрены в разд 2.4.4.
 Схема 2.69
Схема 2.69
|
Тип 2. Параллельные реакции. В примерах, показанных на схеме 2.69, смеси однотипных продуктов образуются благодаря наличию нескольких конкурирующих каналов для данной реакции, и в этих случаях для достижения селективности превращения требуется обеспечить условия для преимущественного (а лучше исключительного) протекания реакции по требуемому направлению. В случае (4) конечный результат превращения зависит, во-первых, от направления атаки Вг4" на тот или иной атом углерода двойной связи, что определяет соотношение позиционных изомеров в образующейся смеси (147+ 148): (149+ 150),а во-вторых, от ориентации подхода нук-леофила НО", определяющей образование продуктов цис- или транс-прн-соединения [соотношение (147+ 149): (148+ 150)).Если селективность образования продуктов присоединения по правилу Марковникова, 147и 148,— это легко достижимая цель, то обеспечение эффективного управления стереохимией присоединения уже относится к категории довольно непростых задач. В сходном случае [(реакция (5)] стереохимия продукта восстановления определяется направлением атаки гидридного реагента по карбонильной группе с одного из двух альтернативных направлений — «снизу» или «сверху» плоскости цикла. Принципы решения этой проблемы обсуждены в разд. 2.4.3.
Тип 3. Последовательно-параллельные реакции. Для примеров, приведенных на схеме 2.70, характерны трудности и первого, и второго типов превращений. Поскольку показанные субстраты полифункциональны, то уже пер-вая из стадий может приводить к образованию смесей продуктов реакций. Доплнительное осложнение обусловлено тем, что каждый из образующихся при этом продуктов, в свою очередь, способен реагировать с тем же реагентом.
Очевидно, что задача обеспечения селективности реакции в таких превращениях существенно сложнее, чем в рассмотренных выше случаях. Действительно, для избирательного получения одного из продуктов первой стадии необходимо не только обеспечить селективное протекание этой реакции по одной из имеющихся групп, но и заблокировать следующие стадии, г. е. обеспечить инертность первичного продукта в условиях реакции. Здесь могут использоваться самые различные приемы. Так, например, при ацети-лировании глицерина [превращение (6)] задача селективного получения мо-но- или бисацетилированных производных по первичной гидроксильной группе (группам) сравнительно легко решается при использовании такого мягкого реагента, как уксусный ангидрид, в требуемых стехиометрических количествах. В то же время моноацетилирование вторичного гидроксила достижимо лишь при условии, что оба первичных гидроксила защищены какой-либо легко удаляемой впоследствии группой (подробнее о принципах использования защитных групп см. разд. 2.4.5.). Дополнительные трудности возникают в тех нередких случаях, когда продукт, получаемый на первой стадии, оказывается более реакционноспособным, чем исходный субстрат. Так, например, при алкилировании толуола по Фриделю—Крафтсу [реакция (7)) присоединение первой алкильной группы резко повышает нуклеофильность ароматического ядра, так что повторное алкилирование протекает быстрее, чем первая стадия. Взаимное влияние функциональных групп является отнюдь не исключением, а правилом, особенно в тех случаях, когда функциональные группы сближены или разделены системой кратных связей. Однако подобное влияние может иметь результатом не только ускорение, но и замедление реакций. Именно этим можно воспользоваться для того, чтобы добиться моноалкилирования толоула. С этой целью вместо аликлирования используют ацилирование, при котором входящая ацильная группа пассивирует ароматическое ядро по отношению к электрофильной атаке. Благодаря этому реакция протекает почти исключительно как монозамещение. Последующее восстановление
 Схема 2.70
Схема 2.70
|
кетогруппы в полученном продукте и даст требуемое моноалкилпроиззодное толуола [реакция (8), схема 2.70].
Рассмотренные типы ситуаций ясно показывают, сколь многогранна и сложна проблема селективности в целом. Вообще говоря, любое органическое соединение полифункционально (даже простейшее из них — метан — образует при хлорировании набор продуктов от СН3Сl до ССl4)- Поэтому неудивительно, что проблема селективности реакций является в действительности ключевой при планировании синтеза.
Из нашего схематического рассмотрения видно, сколь различным может быть характер препятствий на пути к достижению желаемого результата. Соответственно, различны и принципы решения синтетических задач на селективность. Возможность решения задач, связанных с возможностью протекания параллельных реакций (тип 1), в значительной степени обусловлена такими необходимым характеристиками синтетического метода, как его чистота и избирательность. В самом общем виде эти вопросы мы уже обсуждали в предыдущих разделах. Поэтому ниже мы сосредоточим внимание главным образом на задачах, относящихся к типу 2 и — в меньшей степени — к типу 3 по нашей классификации. Речь пойдет главным образом о некоторых принципах решения задач, основанных на вариациях в природе реагента, структуры субстрата и химизма основной реакции. Стоит отметить, что эти пути решения, хотя они и относятся к категории основных, отнюдь не являются единственными. Определенную пользу может принести также и чисто физический приемы, такие, как удаление целевого продукта из равновесной смеси или управление ходом реакции, основанное на понимании кинетических закономерностей конкурирующих процессов [22а].
Теперь несколько терминологических замечаний. Предпочтительное протекание реакций по одной из нескольких родственных, но химически различных функциональных групп сусбетрата, обычно называют хемоселективностью. Если речь идет об избирательности по отношению к определенному положению в молекуле, принято говорить о региоселективности. Если же имеется в виду предпочтительное образование одного из пространствен-(ных изомеров, то пользуются термином стереоселективность. Наконец, гели удается добиться полной селективности, то такой результат характеризуют Термином специфичность (соответственно хемо-, регио- или стерео-). Наконец, существует еше один аспект селективности, связанный с возможностью образования двух оптических антиподов — энантиомеров. Обсуждению общих подходов к решению этой чрезвычайно важной проблемы, а также методологии разработки частных ее решений посвящено несколько десятков монографий и сотни обзоров (см., например, [22Ь]). Как нам представляется, вряд ли было целесообразно пытаться дать содержательное изложение результатов поисков в этом направлении в контексте обсуждения данной главы. С некоторыми общими принципами, применяемыми для решения проблем энантиоселективности, читатель сможет ознакомиться в гл. 4 (см. разд. 4.2.3.3).
Селективность обеспечивается выбором подходящей реакции
В этих случаях речь шла об обеспечении хсмоселективности превращения субстрата. Разберем несколько примеров иного типа, иллюстрирующих возможность… Рассмотрим для начала модельную структуру, диен 151 (схема 2.71), содержащую…Варьирование природы реагентов как способ управления селективностью реакции
В конце 1940-х годов в практику органической химии был внедрен принципиально новый и мощный восстановитель — алюмогидрид лития. Отвлекаясь от… В то же время селективное восстановление альдегидной группы в 156 осуществить… Наконец, если задача состоит в исчерпывающем восстановлении субстра-татипа 156, то можно воспользоваться еще одним, на…Селективная активация
Альтернативных реакционных центров субстрата
Зададимся теперь вопросом: а зачем, собственно, здесь нужна подобного рода… Поэтому в условиях реакции будет также происходит депротонирование ке-тона 171с последующим алкилированием…Защита функциональных групп как универсальный способ управления селективностью реакций
Рассмотрим некий субстрат А—X, для которого хорошо отработан метод его превращения в продукт A-Z. Допустим теперь, что конкретная задача состоит в… Такая маскировка, или защита функций, — прием, чрезвычайно широко используемый… Ранее на этой же системе мы показали, каким образом можно добиться селективного восстановления только формильной…Реагенты, эквиваленты, синтоны
Идеальный органический синтез: фантастика или достижимая цель?
Пофантазируем немного на тему о том, каким бы хотелось видеть идеальный органический синтез (недалекого будущего?). Мы говорили о том, что синтез… Современный конструктор или ученый, работающий в инженерной области,… Именно в таком духе представляется идеальный органический синтез, и к этому ведут современные тенденции его развития.…Синтоны как универсальные (хотя и виртуальные)
Строительные блоки и их реальные синтетические эквиваленты
Говоря выше о синтетическом методе как о «черном ящике», операторе, с помощью которого можно производить строго определенные преобразования структуры исходного соединения, мы пока акцентировали внимание на реакциях, ведущих к такому преобразованию, и мало говорили о тех «строительных блоках», которые можно встроить в собираемую молекулу с помощью того или иного метода. Вот об этих «кирпичиках» и пойдет речь далее.
Реагенты и синтетическая эквивалентность
Понятием «синтетический эквивалент» мы уже неоднократно пользовались. Выражение «синтетическая эквивалентность» подразумевает, что использование… Еще более высокий уровень обобщения достигается при учете возможных… С таким же основанием ацетальдегид может рассматриваться как эквивалент катиона СН3СО+, поскольку продукты его реакций…Понятие о синтонах
В действительности содержание и смысл термина «синтон» проще всего пояснить путем анализа живых примеров, ане с помощью сухих дефиниций. Синтон -СООН. Сочетание такого синтона с каким-либо электрофилом описывается… Е+ + ‑СООН → Е-СООНСинтонный подход как инструмент в разработке путей синтеза
Нередко при ретросинтетическом анализе та или иная разборка может привести к двум фрагментам, один из которых представляет собой хорошо знакомый… Один из вариантов разборки ацетопропилового спирта (236) приводит к…Изоструктурные синтоны обратной полярности
Другим примером пары реагентов, соответствующих изоструктурным синтонам противоположной полярности, являются аллилгалогенид и аллил-силан. Первый из… нуклеофильного синтона СН2=СНСН2. Очевидно, что наличие двух таких доступных… Точно также для введения ароматического остатка синтетик может воспользоваться вариантом, предусматривающим…Построение циклических структур
Специфика задач при синтезе циклических соединений
Вообще говоря, построение молекул, в состав которых входит замкнутая цепь углеродных атомов (цикл), требует решения уже знакомых нам задач… Рассмотрим простейшую модельную ситуацию, когда цикл образуется из… Легко видеть, что в молекулах такого типа взаимодействие между концевыми функциями может протекать не только по схеме…Обычные методы ациклической химии в построении циклических систем
Малые циклы: производные циклопропана и циклобутана
Реакции (1) {28а] и (2) [28Ь] являются примерами хорошо известного сочетания по Вюрцу. Примечательно, что даже в этих случаях, где результатом… В значительной мере сходная ситуация имеет место для циклобутанов с той… Особенно эффективна реакция Вюрца для построения циклобутанового фрагмента в тех случаях, когда структура субстрата…Пят- и шестичленные циклы
Внутримолекулярное взаимодействие енолятного нуклеофила и карбонильного электрофила, приводящее к образованию шестичленного цикла, — хорошо… Столь же высока селективность внутримолекулярной альдольной конденсации 1,4-дикарбонильных соединений (например,…Циклы большего размера. Принципы макроциклизации. Эффекты многоцентровой координации
По мере снижения скорости лактонизации все в большей степени преобладающим становится межмолекулярная конденсация с образованием оли-гомерных… Изменить ход событий в желательном направлении можно, очевидно, двумя…Циклоприсоединение - методы, специально созданные для получения циклических структур
Нетрудно заметить, что все ранее рассмотренные методы циклообразования имеют одну общую особенность: циклизация осуществляется как… Существует, однако, обширный класс методов, основанных на ином топологическом…Циклоприсоединение
Именно благодаря такому эффекту стабилизации переходного состояния последнее оказывается достаточно энергетически выгодным, а потому энергия… В классическом варианте реакции Дильса—Альдера в качестве 4л-компо-ненты…Циклоприсоединение в синтезе производных циклобутана
В случае термически индуцированного процесса согласованный механизм…Синтез циклопропанов путем [2 + 1]-циклоприсоединения
Карбены — крайне нестабильные, реакционноспособные частицы [33а], которые…Селективность циклообразования в комплексах переходных металлов
Ситуация изменилась решающим образом в 1940-х годах, когда Реппе с сотр. [34а] показали, что циклооктатстраен может быть получен с высоким выходом в… Еще в XIX в. было показано, что ацетилен способен претерпевать термическую… Другими словами, высокоупорядоченное и потому энтропийно крайне невыгодное переходное состояние для образования 137из…Радикальные реакции и их роль в синтезе циклических соединений
Типичная схема механизма цепной гемолитической реакции включает стадии инициирования (т. е, генерации радикальной частицы), роста цепи и ее обрыва,… Легко понять, что результат подобной реакции может быть синтетически полезным лишь втом случае, если радикал А,…Расщепление связей С-С и перестройка углеродного скелета как синтетические методы
Расщепление одинарных связей С-С
Пожалуй, наиболее известный и очевидный пример конструктивной роли «деструктивной» реакции — декарбоксилирование алкилированных производных… Разумеется, декарбоксилирование может потребоваться не только в случае…Синтетическое использование реакций расщепления двойной углерод-углеродной связи
Созидательный потенциал реакций, приводящей к разрыву углерод-углеродных связей, еще более наглядно может быть продемонстрирован на примере… Один из самых важных случаев синтетического использования окислительного… Как окислительное расщепление двойной связи, так и альдольная конденсация в системах, подобных 460,относятся к…Перегруппировки углеродного скелета и некоторые возможности их использования в полном синтезе
В числе этих примеров мы видим пинаколиновую перегруппировку (1) и… Открытие скелетных перегруппировок в начале XX в. породило немало проблем, поскольку превращения подобного типа…Перегруппировка Кляйзена-Джонсона—Айрленда и гидрокси-перегруппировка Коупа
Одним из методов получения аллилвиниловых эфиров служит переэтери-фикация алкилвиниловых эфиров аллиловыми спиртами, катализируемая солями ртути.… Превращение аллилвинилового эфира 482в альдегид 483иллюстрирует уникальность синтетического потенциала перегруппировки…Трансформации малых циклов и их роль в полном синтезе
Материал, который относится к этой теме, очень обширен. Ему целиком посвящены четыре выпуска Topics in Current Chemistry [Top. Сшт. Chem.,… На схеме 2.159 представлено два синтеза природных сесквитерпенов, содержащих… Первая стадия синтеза хирсутена (512)[40т] — [2 + 2]-циклоприсоедине-ние циклопентена 513с метилхлоркетеном,…Заключительные замечания
Хотелось бы сделать еще одно замечание более общего характера. Материал, относящийся к методам органического синтеза, огромен по объему, и поэтому…Литература
1. (a) Van Татёеп Е. £., PappasS. P. J. Am. Chem. Soc, 85,3297(1963); (b) Schleyer P. von A, Donaldson M.M. J. Am. Chem. Soc, 82, 4645 (I960).
2. (а) Об истории открытия этой реакции см.: Вепоп J. A. Tetrahedron, 48,3 (1992); (b) Normant К. Adv. Org. Chem., v. 2, p.l, Raphael R. R. (ed.), Interscience Publishers, New York, I960; (c) Pedenen С J. J. Am. Chem. Soc, 89,2495 (1967); (d) обзор см.: Вебер В, П., Токелъ Г. В. Межфазный катализ в органическом синтезе. — М.: Мир, 1980,327 с.
3. О структуре и свойствах различных карбокатионных интермедиатов, см.: Olah G., Schleyer P. von R. Carbonium Ions, vols. 1—5, Wiley, New York, 1968-1976; краткий обзор см.: BetheU G. Reactive Intermediates, v. 1, Wiley, New York, 1978.
4. Bates R. В., Ogle C. A. Carbanion Chemistry, Springer Vertag, New York, 1983; см. также: StowellJ.C. Carbanions in Organic Synthesis, Wiley, New York, 1979.
5. Davies S. G. Organotransition Metal Chemistry: Applications lo Organic Synthesis, Organic Chemistry Series, Baldwin'J. E.(ed.), v. 2, Pergamon, Oxford, 1982; см. также: Cottman J. P., Hegedus L. S., Norton J. R., Finke R. G. Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, CA, 1987.
6. О синтетическом использовании аллильных реагентов см. обзор: Magid R. М. Tetrahedron, 36,1901 (1980).
7. Mori К. Tetrahedron, 30, 3807 (1974).
8. House И. О. Modern Synthetic Reactions, 2nd ed. The Benjamin/Cummings, Memo Park, 1972, ch.9, pp. 492-623.
9,(a) см.: Etcher T. in The Chemistry of Carbonyl Group, Patai S. (ed.) Interscience, New York, 1970, pp.621-693; (b) Imamoto Т., Takiyama N, Nakamura K. Tetrahedron Lett., 26, 4763 (1985); Nagasawa K, Ito K. Heterocycles, Z8, 703 (1989).
10. Cm. [8], ch. 10, pp. 629-733.
11. MasamuneS., Choy W. Aldrichimica Acta, 15,47(1982).
12. (а) Обзоры см.: MaerckerA. Org. React., 14,270 (1965); Bestmann H. J., Vostrowsky 0. Top. Curr. Chsm., 109,85 (1983); (b) Wittig G., Geissler G. Ann., 580,44 (1953); об истории см.: Wittig G. Pure Appl. Chem., 9, 245 (1964); (c) Wadsworth W. W., Jr. Org. React, 25, 73 (1977); (d) Ramirez F, Desai N. В., McKehie N. J. Am. Chem. Soc, 84, 1745 (1962); <e) Corey E. I, Fuchs P. L. Tetrahedron Lett., 1972,3769; см. также: Matsumoto M., Kuroda K. Tetrahedron Lett., 1980, 4021.
13. Обзор см.: Bergman* E. D., GinsburgD., Pappo R. Org. React, 10,179 (1959).
14. (a) Rapson W. S., Robinson R. J. Chem. Soc, 1935,1285; см. также: du Feu E. C, McQuilkn F. J., Robinson R. J. Chem. Soc, 1937,53; (b) см. обзоры: Jung M. E. Tetrahedron, 32, 3 (1976); Gawley R. E. Synthesis, 1976,777; (с) Назаров И. Н., Завьялов С. И. Изв. АН СССР, отд. хим. наук, 1952,300; (d) Tramontini M. Synthesis, 1973,703; (е) см., например, список наиболее употребительных модифицированных акцепторов Михаэля (более десятка наименований), приведенный в монографии: Larock R. С. Comprehensive Organic Transformations, VCH Publishers, 1989,p. 669.
15. (a) Stork G. Pure Appl. Chem., 17, 383 (1968); (b) Taylor R. J. K. Synthesis, 1985,364; (c) Posner G. H. An Introduction to Synthesis Using Organocopper Reagents, Wiley, New York, 1980; (d) Recent Developments in Organocopper Chemistry, Tetrahedron Symposia-inPrint,№35,Tetrahedron,45,349(1989); Posner G.H.Org. React., 19,1 (1972)и22,253 (1975); (e) см. также монографию: PerlmunerP. Conjugate Addition Reactions in Organic Synthesis, Tetrahedron Organic Chemistry Series, Pergamon, Oxford, vol. 9,1992.
16. (a) Normant J. F. AlexakisA. Synthesis, 1981,841; см. также: EnderE. Tetrahedron, 40, 641 (1984); (b) Cahiez G., Alexakis A., Normant J. F. Tetrahedron Lett, 21, 1433 (1980); (c) MarfatA., McGuirk P. A, HelquistP. J. Org. Chem., 44,3888 (1979); (d) KnightD. W., OjharaB. J. Chem. Soc, PerkinTrans. 1,1983,955; см. также: BakerR., BillingtonD. C, Ekanayake N.J. Chem. Soc, Perkin Trans. 1,1983, 1387.
17. (a) Smit W. A. in: Soviet Scientific Reviews, B. Chem., v. 7, 155, Harwood Academic Publishers Gmbh and OPA, Volpin M. E. (ed.), Amsterdam, 1985; (b) Hemming I. Chem. Soc. Rev., 10,83 (1981); (c) Weber W. Silicon Reagents for Organic Synthesis, Springer-Verlag, New York, 1983, Ch. 11, p. 173; (d) Mukaryama T. Org. React, 28, 203 (1982); см. также: Mukaiyama Т., Murakami M. Synthesis, 1987,1043; (e) ReetzM. Г., Maier W. F., Chatziiosifldis I., GiannisA., Heimbach H., Lowe U. Chem. Ber. 113,3741 (1980); обзор см.: Reetz M. T. Angew. Chem., 94, 97 (1982); (0 RajanBabu T. V. J. Org. Chem., 49, 2083 (1984); см. также: Narasaka К., Soai К., Mukaiyama Т. Chem. Lett.,
1974,1223; (g) обзоры см: Reetz M. T. Angew. Chem., Int. Ed. Engl., 23, 556 (1984); Reetz M. T. Ace Chem. Res., 26,462 (1993).
18. (a) Nicholas K. M. Ace Chem. Res., 20, 207 (1987); (b) Schegolev A. A., Smit W. A., Kalyan Y. В., Krimer M. Z., Caple R. Tetrahedron Lett., 23, 4419 (1982); см. также:
Caple R., in: Organic Synthesis: Modern Trends, Chizhov O. S. (ed.), Blackwell Scientific Publications, London, 1987, p. 119; (с) с возможностях использования в полном синтезе см. в обзоре: Smit W. A., Caple Я, Smoiyakova I. P. Chem Rev., 94,
• 2359(1994).
19. (a) Larock R. С. Comprehensive Organic Transformations. A Guide to Functional Group : Preparations, VCH Publishers, 1989; (b) Soloveichik S., Krakauer H. J. Chem. Ed., 43, 532 (1966); см. также монографию: Матье Ж., Панико Р., Вейль-Рейналь Ж. Изменение и введение функций в органическом синтезе. — М.: Мир, 1980; (с) [19а], р.354; (d) Murata S., Suzuki M., Noyori R. J. Am. Chem. Soc, 101,2738 (1979); см. обзоры: Omura K., Swem D. Tetrahedron, 34,1651 (1978); MancusoA. J, Swem D. Synthesis, 1981,165; (e) [19a], p.604; (f) см. ссылки в монографии.: Hudlkky М. Oxidations in Organic Chemistry, ACS Monograph 186, Wasington. DC, 1990; (g) Epstein W. W., Sweat F. W. Chem. Rev., 67, 247 (1967); (h) Corey E. J, Oilman N. W., Ganem B. E. J. Am. Chem. Soc, 90, 5616 (1968); (i) [19a], p. 456; (j) KamkiГ., Sharpless ■ K. B. J. Am. Chem. Soc, 102,5974 (1980); см. также: Hanson R. M., Sharpless К. В. J. Org. Chem., 51,1922 (1986); (k) KursanovD. N, PamesZN, Loim N. M. Synthesis, 1974,633; КурсаиовД. Н., Парте 3. H, Калинкин М. И., Лойм Н. М. Ионное гидрирование. — M.: Химия, 1979; (1) Krishnamurthy S., Brown H. С J. Org. Chem., 47, 276 (1982); примеры использовании триэтилборгидрида лития (супергидрид) как самого эффективного реагента для восстановления как алкилгалогенидов, так и спиртов см.: Bmvn И. С, Krishnamurihy S. J. Am. Chem. Soc, 95, 1669 (1973); Brown H. C, Kirn S. C, KHshnamurthyS. J. Org. Chem., 45,1 (1980); (m) Corey E. J., Achiwa К J. Org. Chem., 34,3667 (1969); (n) Adlington R. M., Barrett A. G. M. Ace. Chem. Res., 16, 55 (1983).
20. (a) Graham J. С Tetrahedron Lett., 1973,3825; (b) Gilbert K. E, Borden W. T. i. Org. Chem., 44,659 (1979); (c) Jacobson R. M. Tetrahedron Lett., 1974, 3215; (d) Abael-Magid A. F, Maryanoff С A., Carson K. G. Tetrahedron Lett., 31, 5595 (1990); (e) Sharma D. N, Shama P.P. Tetrahedron Lett., 26, 2561 (1985); (0 Jung M. E, Lyster M. A. 1. Org. Chem., 42, 3761 (1977); обзор см: Olah G. A.. Nahang S. С Tetrahedron, 38,2225 (1982).
21. (a) WillstatterR., Waser E. Ber., 44,3423 (1911); (b) Bochkov A. F., Zaikov G. E. Chemistry of the O-Glycosidic Bonds: Formation and Cleavage, Pergamon Press, Oxford, 1979; Бочков А. Ф., Афанасьев В. А., Занков Г. Е. Образование и расширение гликозидных связей. — М.: Наука, 1978.
22. (а) Бочков А. Ф. ЖОХ, 19, 1654 (1983); (b) kumi Y, Tai A. Stereo-differentiating Rections, Academic Press, New York, 1977; Morrison J. D. Asymmetric Synthesis, Academic Press, New York, 1983/84, v. 1-5.
23. (a) 06 истории и общих аспектах гидридного восстановления см. обзор: Brown Н. С, Krishnamunhy S. Tetrahedron, 35,567 (1979) и монографии: Хайош А. Комплексные гидриды в органической химии. — Л.: Химия, 1971 и Мигович В., Михайлович М. Алюмогидрид лития и его применение в органической химии. — М.: ИЛ, 1957; (b) Hutchins R. О., Kandasamy D., Maryanoff С. A., Masilamani D., MaryanoffB. E. J. Org. Chem., 42, 82 (1977); (с) Adams С. Synth. Comm., 1984,1349;
(d) см., например: СгаЪЫ P., GuzmdnA., Vera M. Tetrahedron Lett, 1973,3021; (e) SarkarD. C, DasA.R., Ranu В. С J. Org. Chem., 55,5799 (1990); (0 Fisher G. В., Fuller J. C, Harrison, J., Alvarez S. G, Burkhardt E. R, Gorolski С. Т., Singaram В. J. Org. Chem., 59, 6378 (1994); (g) см., например: Tsuda Т., Yazawa Т., fValanabeK., Fujii Т., Saegusa T.J. Org. Chem., 46, 192 (1981); (h) Burgstahler A. W., Sanders M. E. Synthesis, 1980,400; (i) Brown H. C, Krishnamurthy S. Aldrichimica Acta, 12, 3 (1979); (j) Парнес 3. M., Лойм Н. М., Баранова В. А., Курсанов Д. Н. ЖОрХ, 7, 2066 (1971).
24. (а) См. например: Hoehn W. M., Moffett R. В. J. Am. Chem. Soc, 67, 740 (1945); (b) Friour G., AlexakisA., Cahiez G., Normant J. F. Tetrahedron, 40, 683 (1984); Friour G, Cahiez G., NormcntJ. F. Synthesis, 1985, 50; (c) Cahiez G., Rtvas-Enterios J., Gragner-Veyron H. Tetrahedron Lett., 27, 4441 (1986); (d) LipshulzB. H. Synthesis, 1987,325;
(e) Oilman H, StrakyJ. M. Rec. Trav. Chim., 55,821 (1936) и цитированные в этой статье работы; (f) House Н. О., Respess G. О., Whitesides G. M. J. Org. Chem., 31,3128
• (1966); (g) HouseH. О. Асе. Chem. Res., 9,59 (1976); (h) Posner G. Org. Reactions, 19, 1 (1972); (i) см., например: Corey E. J., Posner G. J. Am. Chem. Soc, 89, 3911 (1967); 0) Corey E. J., Posner G. J. Am. Chem. Soc, 90, 5615 (1968); (k) Achyutha Rao S., Knochel P. J. Org. Chem., 56,4591 (1991): (1) Kotsuki H, Kcdota L, Ochi M. J. Org. Chem., 55,4417 (1990); (m) Harris Т. М., Harris С. М. Organic reactions, 17, 155 (1969).
25. (a) Brownbridge P. Synthesis, 1983,1; там же, 1983,85; (b) Fleming L, Paterson I. Synthesis, 1979,736; (с) см., например, методрегио- истереоселективного алкшш-рования кегонов с использованием литиевых производных М,М-диметилгидразо-нов: Corey E.J., Enders D. Tetrahedron Lett., 1976,1;там же, 1976,l;(d)SckreiberS. L, WangZ. J. Am. Chem. Soc, 107,5303 (1985).
26. (а) Одинокое В. Л., Бакеева Р. С, Галеева Р. И., Ахунова В. Р., Мухтаров Ю, Г., Тол-стиков Г. А., Халилов Л. М,, Панасенко А. А. ЖОрХ, 15, 2017 (1979); (B) см., например: Bosch M. P., Camps J., CoilJ., Guerrero A., Tatsuoka Г., MeinwaldJ. J. Org. Chem., 51, 773 (1986); (c) Greene T. M., Wuts P. G. M. Protective Groups in Organic Synthesis, 2nd ed., J. Wiley, New York, 1991; (d) Kocienski P. J. Protecting Groups, G. Thieme Verlag, Stuttgart, 1994; (e) Mcolaou K. C, Claremon D. A., Barnette W. E. J. Am. Chem. Soc, 102,6611 (1980); (0 JungM. E., SpeltzL. M J. Am. Chem. Soc, 98,7882 (1976); (g) Keinan E., Sahai M., Shvity R. Synthesis, 1991,641; (h) Mpango G. B, Mahala-banis K, K., Mahdavi-Damghani Z, Snieckus V. Tetrahedron Lett., 21, 4823 (1980); (i) Knapp S., CalienueJ. Synth. Commun., 10, 837 (1980).
27. (a) Corey E. J. Pure Appl. Chem., 14,19 (1957); (b) Corey E. J., ChengX. M. The Logic of Organic Sytnhesis, Wiley, New York, 1989; (с) Марч Дж. Органическая химия. В 4-х т. Т. 2, с. 214. — М.: Мир, 1988; (d) Casey С. P., Marten D. F. Synth. Commun., 3, 321 (1973); (е) сжатое изложение идеологии подхода см.: Seebach D. Angew. Chem., Int. Ed. Engl, 8, 639 (1969); см. также: Lever О. W., Jr. Tetrahedron, 32,1943 (1971); (0 Hose T.A. Umpoled Synthones, Wiley, New York, 1987; (g) впечатляющий пример использования подхода для сборки несимметричных 1,4-дикетонов см.: Hermann L, RichmanJ. E., Schlessinger R. H.Tetrahedron Lett., 1973,3271; (h) обзор см.: Grobel B.-T., Seebach D. Synthesis, 1977,357; (i) Seebach D., Sewing В., Kalinowski H.-O., Lubosch W., Renger B. Angew. Chem., 89, 270 (1977); 0) Corey E. J., Kozikovsky A. P. Tetrahedron Lett., 1975,925; см. также: Seebach D., Kolb M. Ann., 1977,811; Seebach D., Bilrstinghaus R., Grobel B.-T., Kolb M. Lieb. Ann. Chem., 1977,830; (k) Chou T.-S., Knochel P. J. Org. Chem., 55,4791 (1990); (1) подробный обзор по гетерозамещенным металлоорганическим производным см.: Krief A. Tetrahedron, 36, 2531 (1980); (m) Hose Т. A., KoskimiesJ. К. Aldrichimica Acta, 14,73 (1981); (n)StowellJ. С. Chem. Rev., 84,409 (1984).
28. (a) Wiberg К. В., Lampman G. M. Tetrahedron Lttt., 1963, 2173; (b) Applequist D.E., Fanta G. F., Henrickson B. W. J. Org. Chem., 23,1715 (1958); (с) см. также обзор:
МенчиковЛ. Г., Нефедов О. М. Усп. хим., 63,471 (1994); (d) MisumiA., Iwanaga К., Furuta К., Yamamoto H. J. Am. Chem. Soc, 107,3343 (1985); (e) [8], p. 542; (f) Brewer P. J)., Tagat J., Hergmeter С A., Helquist P. Tetrahedron Lett., 1977,4573; (g) Whitesides G. M., Gutomki F. D. J. Org. Chem., 41, 2882 (1976).
29. (a) Smith, M. B. Organic Synthesis, McGraw-Hill, Inc., 1994, p. 889; (b) см.обзор: Hudlicky Т., Price J. D. Chem. Rev., 89,1467 (1989); о многочисленных примерах
применения методов циклопентаноаннелирования в синтезе см. обзор: Paquette L.
A. Top. Current Chem., 119,1 (1984); (с) Herrmam J. L, Richman J. E., Schlessinger R.
H. Tetrahedron Lett., 1973,3275; (d) Mundy B. P., WilkeningD., Lipkowitz К. В. J. Org. Chem., 50,5727 (1985); см. также: Furuta К., MisumiA., Mori A., Ikeda N., Yamamoto
H. Tetrahedron Lett., 25,669 (1984); (e) Piers E., Karuncratne V. J. Chem. Soc, Chem.
Commun., 1983,935; (f) Bunce R. A., Wamsley E. J., Pierce J. D., Shellhammer A. J., Jr.,
Drumright R. E. J. Org. Chem., 52,464 (1987); см. также: Yamaguchi M., Tsukamote M.,
Hirao 1. Tetrahedron Lett., 26,1723 (1985); (g) см. также: Назаров И. Я., Зарецкая И.
И. Изв. АН СССР, отд. хим. наук, 1944, 65; (h) Santelli-Rouvier К, SantelH M.
Synthesis, 1983,429; (i) обзор см.: Trosl В. Angew. Chem., Int. Ed. Engl, 25, 1 (1986).
30. (а) Подробное обсуждение роли энтропийных и энталышйных факторов см.: DeTarD. F., LuthraN. P. J. Am. Chem. Soc, 102,4505 (1980); (b) March J. Advanced Organic Chemistry, 3rd. edn., Wiley, New York, 1985, p. 184; (c) Tiegler K. inMethoden der Organischen Chemie (Houben-Weil); Muller E. (ed.), George Thieme Veriag, 1955, vol. 4/2; см. также обсуждение в ссылке [29а]. р. 6 И ив обзоре: Прелог В. в кн. Перспективы развития органической химии, Тодд А. (ред.). — М.: ИЛ, 1959, с. 77; обзор по препаративным методам синтеза макролидов см.: Nicolaou К. С. Tetrahedron, 33,683 (1977); Masamune S., Bates G. S., Corcoran J. W. Angew. Chem., Int. Ed. Engl., 16,585 (1977); (e) Corey E. J., Nicolaou К. С J. Am. Chem. Soc, 96,5614 (1974); обзор см.: Mukaiyama Т. Angew. Chem., Int. Ed. Engl., 18, 707 (1979); краткий обзор см.: MukerJ. Angew. Chem., Int. Ed. Engl., 30,1452 (1991); (0 Corey E. J., Ulrich P., Fitzpatrick J. M. J. Am. Chem. Soc., 98,222 (1976); об альтернативном подходе, основанном на образовании смешанного ангидрида 2,4,6-трихлорбензой-ной кислоты и со-оксикислоты см.: Hikota М., Топе Н, Horita К., Yonemitsu О. J. Org. Chem., 55,7 (1990) и цитированные в этой статье работы; (g) Yamamoto H., Maruoka К. J. Am. Chem. Soc, 103,6133 (1981).
31. (а) Онищенко А. С. Диеновый синтез. — M.: Наука, 1964; (b) Wollweber H. Diels-Alder Reaction, Thieme, Stuttgart, 1972; (c) Woodward R. В., Hoffmann R. Angew. Chem., Int. Ed. Engl., 8, 781 (1969); см. также: Джиякрист Т., Сторр Р. Органические реакции и орбитальная симметрия. — М.: Мир, 1976; (d) Destmoni С, Tacconi G., Barco A., PoUini G. P. Natural Product Synthesis Through Pericyclic Reactions, ACS Monograph 180, ACS, Washington D. C, 1983, ch.5, p. 119; (e) Danishefiky S. Ace. Chem. Res., 14, 400 (1981); (0 WattD. S., Corey E. J. Tetrahedron Lett,1972, 4651; (g) см., например, использование этого подхода в синтезе резер пина: Martin S. F., RiiegerH., Williamson S. A., Grzejszczak S.J. Am. Chem. Soc, 199, 6124 (1987); (h) DanishefskyS., Etheredge S. J. J. Oig. Chem., 44, 4716 (1979); (i) Ca-дых-Заде С. И., Петров А. Д. ЖОХ, 28, 1591 (1958); Carter М. /., Fleming I. J. Chem.Soc, Chem. Comm., 1976, 679; (j) CarrZ V. C, Williams R V., Paquette L. A. J. Org. Chem., 48,4976 (1983); (k) Bonjouklian R, Ruden R. A J. Org. Chem., 42,4095 (1977); (1) Corey E. J., Weinshenker N. M., ShaqfT. K., Huber W. J. Am. Chem. Soc., 91, 5675(1969); (m) Masamune S., Cuts H, Hogben M. G. Tetrahedron Lett., 1966, 1017; (n) Eaton P. E., Or Y. S, Branca S. J. J. Am. Chem. Soc., 103, 2134 (1981); (о) обзоры см.: Oppoker W. Angew. Chem., Int. Ed. EngL, 16, 10 (1977); Brieger G., BennelJ. N. Chem. Rev., 80, 63 (1980); (p) Brieger G., Anderson D. R. J. Org. Chem., 36, 243 (1971); (r) HouseH. 0., Cronin T. H. i. Org. Chem., 30, 1061 (1965); (s) Nicolaou K. C, Li W. S. J.Chem. Soc., Chem. Comm., 1985,421; (t)StorkG., Chan Т. У J. Am. Chem. Soc, 117, 6595 (1995) и цитированные в этой статье работы; (u) Oikawa Н, Yokota Т., Abe Т., IchiharaA., Sakcmura S., Yoshizawa Y., Vederas J.C.J. Chem. Soc, Chem. Comm., 1989, 1282; Oikawa H., Yokota Т., khihara A., Sakamura Д., там же: 1989, 1284; (v) Oikawa H, Suzuki Y, NayaA., Katayama X., khihara A. J. Am. Chem. Soc, 116, 3605 (1994); (w) khihara A., Afiki M., Tazdki H., Sakamura S, Tetrahedron Lett., 28,1175(1987).
32. (a) Cm. [31d], Ch. 3, p.33; (b) Brady W. Г., PatelA. D. J. Org. Chem., 38, 4106 (1973); обзор см.: Brady W. T. Tetrahedron, 37, 2949 (1981); (c) Springer J. P., Clardy J., Cole R. J., Kirksey J. W., Hill R. K, Carlson R. M., IsidorJ. L.J. Am. Chem. Soc, 96, 2267(1974); (d) Snider B. B. Chem. Rev., 88, 793 (1988); (e) Bisceglia R. H, Cheer С J. J. Chem. Soc, Chem. Comm., 1973, 165; (f) Snider В. В., Hui R. A. H. F. J. Org. Chem., 50, 5167 (1985); (g) обзоры см.: Eaton P. £. Ace. Chem. Res., 1, 50 (1967); De Mayo P. Ace. Chem. Res., 4, 41 (1971); (h) Corey E. J., Mitra А В., Uda H. J. Am. Chem. Soc, 86,485 (1964); (i) см.: примеры в работе [31d], ch. 3, p.42; (j) обзор см.: Crimmins M. T. Chem. Rev., 88, 1453 (1988); (k) BeckerD., Haddad N. Tetrahedron Lett., 27, 6393 (1986); (1) WolffS., Agosta W. С J. Am. Chem. Soc, 105, 1292(1983); (m) Oppolzer W. Ace. Chem. Res., 15,135 (1982).
33. (а) См. краткое изложение общих вопросов строения и свойств карбенов в монографиях: Нефедов О. М., Иоффе А. И., Мечников Л. Г. Химия карбенов. — М.:Химия, 1990; Кирмсе В. Химия карбенов. — М.: Мир, 1966, а также в книге: Марч Дж. Органическая химия. В 4-х т. Т. 1, с 249-253. — М.: Мир, 1988; (b) Nefedm О М., Maltsev А. К, Mikaefyan R. С Tetrahedron Lett,, 1971, 4125; (с) Jones M., Jr. Асе. Chem. Res., 7, 415 (1974); (d) Кирмсе В. Химия карбенов. — М.: Мир, 1966, 324 с; Нефедов О. М., Иоффе А. И., МенчиковЛ. Г. Химия карбенов. — М.: Мир, 1990, 373 с; (е)обзорысм.: ТомшовЮ. В., ДокичевВ. А.,ДжемилевУ. М.; Нефедов О М. Усп. хим., 62, 847 (1993); Doyle M. P. Chem. Rev., 86, 919 (1986); (f) Simmons H. E., Cairns Т. L., Vladuchick S. A., Hoiness С M. Org. React, 20,1 (1973); (g)обсуждение общих аспектов препаративной химии карбеноидов, генерируемых из а-галогенметаллоорганических производных, см.: Нефедов О.М., Дьяченко А. И.; Прокофьев А. К.Усп. хим., 46,787 (1977); (h) Campbell J. G. M., Harper S. И. J. Chem.Soc, 1945, 283; (i) Skatteb0lL. J. Org. Chem., 31, 2789 (1966); (j) Щ O. P., Bhatia M. S., Gupta K. C, Malta K. L. J. Indian Chem. Soc, 46, 991 (1969).
34. (a) Reppe W., Schlichting O., Klager K., Toepel T. Lieb. Ann. Chem., 560, 1 (1948);(b) Schrauzer G. N., GiocknerP, EichlerS. Angew. Chem., Int. Ed. Engl., 3, 185 (1964); обсуждение общих проблем ииклоолигомеризации ацетилена в присутствии комплексов переходных металлов, см.: Maitlis, Р. М. }. Organomet. Chem., 200, 161 (1980); (c)AmettE. M., BollingerJ. M.i. Am. Chem. Soc, 86,4729 (1964); (d)Vollhardt К. P. Ace. Chem. Res., 10,1 (1977); (e) обзоры см.: Pauson P. L Tetrahedron, 41, 5860 (1985); Schore N. £ Organic Reactions, 40, 2 (1991); If) Schore N. E, Croudace M. С J. Org. Chem., 46, 5436 (1981); более недавние работы по селективности и механизму реакции см.: KrafflM. Е., Scott I. L, Romero R. H, Feibelman S., Van Pelt С. Е. J. Am. Chem. Soc, 115, 7199 (1993); (g) Shambayati S., Crowe W. E., Schreiber S. L. Tetrahedron Lett., 31, 5289 (1990); (h) Smit W. A., Gybin A- S., Strychkov Y. Т., Mikaelian G. S., CapleR., Swanson E. D.Tetrahedron Lett., 27, 1241 (1986); (i) Schreiber S. L, Sammakia T, Crowe W. E. J. Am. Chem. Soc, 108, 3128 (1986); см. также: Januson Т. F, ShambayatiS., Crowe W. E., Schreiber S. L. J. Am. Chem. Soc, 116, 5505(1994); (j) обзор см.: mike G. J. Organomet. Chem., 200, 349 (1980); (k) см., например: Джемилее У. М., Иванов Г. Е., Толстиков Г. А. ЖОрХ, 11, 1636 (1975); (1) Brenner W., Heimbach P., Wilke G. Ann., 727,194 (1969); (m) обзор см.: Толстиков Г. А., Джемипев У. М., Хим. гетероцикл. соед., 1980,147; (п) многочисленные примеры, иллюстрирующие достижения и перспгктивы применения комплексов переходных металлов как катализаторов в синтезе самых различных по структуре соединений, приведены в обзоре: Frost В. М. Angew. Chem., Int. Ed. EngL, 34, 259 (1995).
35. (а) Обзоры см.: Walling С, Huyser E. S. Org. React., 13, 91 (1963); Vogel H. H. Synthesis, 1970, 99; получение ацеталя левулинового альдегида см.: Mondon A. Angew. Chem., 64, 224 (1952); (b) Jaspers С. P., Curran D. P., Fevig T. L. Chem. Rev., 91, 1237 (1991); (c) Corey E. J., Kang M.-C. J. Am. Chem. Soc, 106, 5384 (1984); (d) Stork G., Mook R., Jr., Bitter S. A., Rychnovsky S. D. J. Am. Chem. Soc, 105, 3741(1983); (e) Srikrishna A., Sharma G. V. R. Tetrahedron Lett., 29, 6487 (1988); (f) Stork G, SherP. M., Chen H.-L. i. Am. Chem. Soc, 108, 6384 (1986); см. также: Keck G. E, Burnett D. A. J. Org. Chem., 52, 2958 (1987).
36. (а) См., например: Wiberg К. В., Lowry В. R., Colby Т. Н. J. Am. Chem. Soc, 83, 3998 (1961); (b) Scheldon Д. A., Kochi J. К Organic Reactions, 19, 279 (1972); см. также: Сергучов Ю. А., Белецкая И. П. Усп. хим., 49, 2257 (1980); (с) Kochi J. К, Bacha J. D. J. Org. Chem., 33, 2746 (1968); (d) Bacha J. D., Kochi J. K. Tetrahedron, 24, 2215 (1968); (e) см. типичные примеры в обзоре: De Lucchi О., Modena G. Tetrahedron, 40,2585(1984).
37. (a) Hassal С H. Org. React., 9, 73 (1957); (b) Starcher P. S., Phillips B. i. Am. Chem. Soc, 80, 4079 (1958); (c) Emmons W. D., Lucas G. B. J. Am. Chem. Soc, 77, 2287 (1955); (d) Payne G. B. Tetrahedron, 18, 763 (1Э62); (e) Martinez. G. R., GriecoP. A., Williams E, Ken-ishi K., SrinivasanС V. J. Am. Chem. Soc, 104, 1436 (1982).
38. (a) Dryhurst G. Periodate Oxidation of Diols and Other Functional Groups, Pergamon Press, Oxford, 1970, p. 191; (b) обзор см.: PerlinA. S. Adv. Carbohydr. Chem. 14, 9 (1959); (c) Schmid С R., Bryant J. D. Org. Synth., 72, 6 (1995); (d) обзор см.: Schroeder M. Chem. Rev., 80, 187 (1980); (e) Corey E. J., NozoeS. J. Am. Chem. Soc, 85, 3527 (1963).
39. (a) Bailey P. S. Ozonation in Organic Chemistry, vol. 1 and 2, Acad. Press, New York, 1978, 1982; см. также: Одинокое В. Н, Толстиков Г. Л. Усп. хим., 50, 1207 (1981); (B) Одинокое В. К, Ахунова В. Р., Бакеева Р. С, Галеева Р. И, Семеновский А. В., Моисеенков А. М., Толстиков Г. А. ЖОрХ, 13, 532 (1977); (с) Corey E. J, Katzenellenbogen J. A., Oilman N. W., Roman S. A., Erickson B. W. J. Am. Chem. Soc,90,5618(1968).
40. (а) См. ссылку [31d], ch. 7, p. 267; о некоторых аспектах синтетического применения перегруппировок см. обзор: Martin S. F. Tetrahedron, 36, 419 (1980); (b) Ziegler F. E. Chem Rev., 88, 1423 (1988); (c) Burgstahler A. W., Nordin I. C. !. Am. Chem. Soc, 83,198 (1961); (d) Johnson W. S., WerthemannL., Bartlett W. R., Brocksom Т. /., Lee T.-T., Faulkner D. J., Petersen M. R. J. Am. Chem. Soc., 92, 741 (1970); (e) см., например: Johnson W. S., Gravestock M. В., McCany B. E. J. Am. Chem Soc 93 4332 (1971); (f) Ireland R. £., Mueller R. H. J. Am. Chem. Soc., 94, 5897 (1972) (g)'Ireland R. R, MuellerR. H., WillardA. K. J. Am. Chem. Soc., 98, 2868 (1976); см также- Wilson S. R., MyersR. S. J Oig. Chem., 40,3309 (1975); (h) см. [31d], ch. 7, p. 279; (i) Evans D. A GolubA. M. J. Am. Chem. Soc., 97, 4765 (1975); см. также: Wilson S. A, Mao D. T. Tetrahedron Lett., 1977,2559; (j) Paquette L. A. Angew. Chem., Int. Ed. Engl 29 609 (1990) (k) Still W. С J. Am. Chem. Soc., 101,2493 (1979); (1) Schreiber S. L, Santini С J Am' Chem. Soc., 106,4038 (1984); (m) Green A. E. Tetrahedron Lett. 21, 3059 (1980); (n) Tobe Y. Yamashita Т., KakiuchiK., Odaira Y.i. Chem. Soc., Chem. Commun., 1984,1259- см также обзор: Bellus D., Ernst B. Angew. Chem., Int. Ed. Engl., 27, 797 (1988); (o) Begley M. J. Pattenden G., Robertson G. M. J. Chem Soc, Perkin Trans. 1,1988,1085 и цитированные в этой статье работы; (р) WongH. N. С, Hon M.-Y., Tse C.-W., Yip Y.-C. TankoJ. Hudlicky Т. Chem. Rev., 89,165 (1989); (r) Mehta G., Ready D. S., MurtyA. N. J. Chem, Soc., Chem. Comm., 1983,824; (s) обзор см.: Kende A. S. Org. React., П, 261 (1960); (t) BarborakJ. С Walts L, PettitRJ. Am. Chem. Soc., 88,1328 (1966); см. также: Eaton P.E. Cole T. W. Jr. J Am. Chem. Soc, 86, 962 (1964); там же: 86,3157 (1964).
* Приведенное соотношение не является просто гиперболой. Соотношение 1 : 1000 считается вполне приемлемым в работах, направленных на создание биологически активных соединений. В последнее десятилетие это положение начинает меняться в лучшую сторону благодаря разработке принципов молекулярного дизайна, о чем пойдет речь в главе 4.
* Отметим, что на протяжении более чем полувека так и оставался нерешенным вопрос о строении димерного углеводорода, которому первоначально было приписано строение гексафенилэтана. Лишь в 1968 г, было строго доказано (с помощью метода ЯМР), что полученный Гомбергом углеводород на самом деле имеет хиноидную структуру 65б [31с]. «Настоящий» гексафенилэтан (65) так и не получен до сих пор [31d].
*Это предположение не столь гипотетично, как может показаться читателю, особенно молодому. Достаточно напомнить, что в 60-х годах усилиями некоторых ученых-чиновников Академии наук СССР работы по полному синтезу были отнесены к разряду второстепенных, что безусловно и явилось одной из причин все нарастающего отставания отечественной органической химик от мирового уровня.
*Значимость этого фактора состоит не только в общеизвестных проблемах, связанных с загрязнением окружающей среды вредными веществами. Не меньшее значение имеет сам факт появления на нашей планете многочисленных искусственно созданных веществ, что может абсолютно непредсказуемым образом сказаться на эволюции биосферы. Частным примгром, иллюстрирующим возможную роль этого нового антропогенного фактора естественного отбора, может служить хорошо известный феномен ускоренного формирования новых резистентных штаммов микроорганизмов, которое индуцируется появлением все новых и новых лекарственных средств.
** Нас могут упрекнуть в том, что мы умалчиваем о новых подходах, появившихся благодаря риработке самых разнообразных и эффективных компьютерных программ для решения задач типа «структура— свойство». Безусловно, использование этих программ может существенно сократить область эмпирического поиска и тем самым ускорить решение таких задач. Хотелось бы, однако, отметить, что эффективность этого подхода в конечном счете более всего зависит от полноты и представительности обрабатываемого банка данных, получаемых все тем же «старым добрым» способом, т.е. путем эксперимента.
* * К настоящему времени уже получено около 15 млн. органических соединений. Темы расширения этого «ассортимента» (примерно 500 новых соединений в день) пока не обнаруживает тенденции к снижению.
* Дальнейшее развитие работ в области дотированных полиацетиленов оказалось чрезвычайно успешным, свидетельством чего может служить присуждение Нобелевской премии по химии за 2000 г. А. Хигеру, А. Мак-Диармиду и X. Сиракаве за «Открытие и развитие области электропроводящих полимеров» (подробнее об этом можно прочесть в статье: Кобрянский В. М., Природа, № 1,с. 7, 2001).
** Уместно заметить, что полианилин был получен еще в 1862 г., но до недавнего времени никто и не подозревал о его довольно высокой электропроводности (200 См/см).
*Сказанное вовсе не означает, что авторы настаивают на научной значимости любой работы, результатом которой является синтез нового вещества. Именно отмеченная выше бесконечность многообразия возможных структур органических соединений заставляет особенно требовательно относится к выбору области синтетического поиска, и исследование в этой области может считаться оправданным только при условии четкой формулировки его цели.
* Бели бы это было иначе и целевые соединения отвечали термодинамическому рагнове-t сию, то проблемы синтеза практически не существовало, ибо в таком случае его можно было .осуществить, используя любой способ (например, нагревание), способствующий приведению системы в состояние равновесия.
*В схеме показано лишь образование п-бромтолуола. В действительности в этой, какивдру-гих подобных реакциях наблюдается образование по такому же механизму некоторого количества о-изомера, что, однако, не меняет общей логики нашего анализа.
* Под перициклическими понимают реакции, идущие через циклическое переходное состояние по согласованному механизму, т. е. одновременным разрывом старых и образованием новых связей.
* Энергия образования такой связи, измеренная для реакции в газовой фазе, составляет 54,3 Дж/моль — величина, весьма солидная для химической реакции.
*Характерной особенностью структуры изопреноидов, определяющей их как четко очерченныйкласс природных соединения, является то, что их молекулы могут быть построены из двухили более остатков изопрена, связанных тем или иным образом в зависимости от пути их биосинтеза, с последующими, довольно прихотливыми модификациями первоначально образующегося ациклического скелета
** Уместно отметить, что биосинтез ациклических изопреноидов также протекает по схеме, вполне адекватно описываемой как сочетание аллильного катиона с аллильным анионом. Разумеется, конкретная природа соединений, служащих эквивалентами электрофилов и нуклеофи-лов в биохимической реакции, достаточно сильно отличается от того, что мы исполыуем в лабораторной практике.
* В строгом смысле термин «реакция Гриньяра» подразумевает использование магнийорга-нических реагентов. По типу реакций Гриньяра реагируют и литийорганические производные, и к ним также относится все сказанное выше о магнийорганических соединениях. Поэтому в синтетическом смысле эти два гипа реагентов почти эквивалентны.
* Эту задачу во многих случаях решают с использованием селектиивных реакций, осуществляемых микроорганизмами, однако мы не имеем возможности рассматривать эти реакции в данной книге.
* Вообще говоря, повлиять она, конечно, может, но отсутствие существенного влияния природы защитной группы на ход последующих реакций является одним из требований к выоору защиты, пригодной для планируемого синтеза.
* На схеме использован символ о, который мы вводим для обозначения отношений эквивалентности между реагентом и синтоном (в математической логике этот символ означает эквивалентность утверждений).
– Конец работы –
Используемые теги: Органический, Синтез0.058
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: ОРГАНИЧЕСКИЙ СИНТЕЗ
Что будем делать с полученным материалом:
Если этот материал оказался полезным для Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.154 сек.
Новости и инфо для студентов