рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Биология
- /
- Генетика, Наследственность, Изменчивость
Реферат Курсовая Конспект
Генетика, Наследственность, Изменчивость
Генетика, Наследственность, Изменчивость - раздел Биология, Введение Генетика (От Гречес. Сепеисоз - «От...
ВВЕДЕНИЕ
Генетика (от гречес. Сепеисоз - «относящийся к происхождению») наряду с морфологией, физиологией и биохимией - является теоретическим фундаментом современной медицины. Предметом генетики человека служит изучение наследственности и изменчивости у человека на всех уровнях его организации и существования: молекулярном, клеточном, организменном, популяционном, биогеохимическом.
Наследственность - свойство организмов обеспечивать материальную и функциональную преемственность между поколениями, а также специфический характер индивидуального развития в определенных условиях среды.
Изменчивость - это свойство организмов изменять наследственные задатки, приобретать новые свойства или утрачивать прежние, реагировать на воздействия факторов среды морфофизиологическими изменениями.
Медицинская генетика исследует больного человека, его больных и здоровых родственников; Изучает генетические механизмы наследственных заболеваний, роль генетических факторов в развитии ненаследственных форм патологии, а также разрабатывает способы их диагностики, профилактики и лечения.
Медицинская генетика составляет важнейшую часть теоретической медицины, рассматривает в связи с патологией следующие вопросы:
1) какие наследственные механизмы поддерживают гомеостаз организма и определяют здоровье индивида:
2) каково значение наследственных факторов (мутации или сочетание определенных аллелей) в этиологии болезней:
3) каково соотношение наследственных и средовых факторов в патогенезе болезней:
4) какова роль наследственных факторов в определении клинической
картины болезней (и наследственных, и ненаследственных);
5) влияет ли наследственная конституция на процесс выздоровления человека и исход болезни;
6) как наследственность определяет специфику фармакологического и других видов лечения
Для врача образование по медицинской генетике включает в себя основы общей генетики (менделизм, учение о хромосомах, химические основы наследственности), основные положения генетики человека (человек как объект генетического исследования) и клиническую генетику.
Клиническая генетика - прикладной раздел медицинской генетики, т.е. применение достижений последней для решения клинических проблем у пациентов или в их семьях.
Клиническая генетика значительно преуспела в своих достижениях с тех пор, как в середине 70-х годов в молекулярной биологии и генетике нашли применение новейшие технологии в исследованиях структуры и функции
ДНК. Классические направления генетики пополнились новым направлением, получившим название обратная генетика, или генетика позиционного клонирования (выделение, клонирование и изучение первичной структуры генов без предварительного знания их белковых продуктов). Для современной клинической генетики характерно широкое применение цитогенетического и молекулярно-генетического анализа семей с наследственной патологией, а также картирование генов. В рамках Международной программы «Геном человека», рассчитанной на 1989-2005гг. ведется тотальное генетическое и физическое картирование генов (определение взаимного расположения генов в молекуле ДНК и расстояние между генами) и секвенирование (определение порядка расположения оснований - нуклеотидов в молекуле ДНК или РНК, или определение порядка аминокислот в белке).
ЗНАЧЕНИЕ ГЕНЕТИКИ ДЛЯ МЕДИЦИНЫ
Прогресс в развитии медицины и общества приводит к относительному возрастанию доли генетически обусловленной патологии в заболеваемости, смертности, социальной дизадаптации (инвалидизации).
По данным каталога МакКьюсика (МсКшюк) в настоящее время насчитывается около 5 тысяч наследственных синдромов (4860). Около 5-5.5% детей рождаются с наследственными или врожденными болезнями.
Известно более 100 нозологических единиц хромосомных болезней (ХБ) и около 900 типов хромосомных нарушений у человека. На долю мультифакториальных болезней (МФБ) приходится 92-93% всех случаев хронической неинфекционной патологии.
Половина спонтанных абортов обусловлена генетическими причинами. Не менее 30% перинатальной и неонатальной смертности связано с врожденными пороками развития и наследственными болезнями с другими проявлениями
Тип и распространенность наследственной патологии у детей:
Тип патологии Распространенность. %
Генные болезни 1 (среди новорожденных)
Хромосомные болезни 0.5 (среди новорожденных)
Болезни с наследственной 3-3.5 (среди детей до 5 лет)
предрасположенностью
Генетические соматические неизвестна
нарушения
Несовместимость матери и 0.4 (среди новорожденных)
плода
Не менее 25% всех больничных коек занято пациентами, страдающими болезнями с наследственной предрасположенностью.
Знание основ медицинской генетики позволяет врач}' понимать механизмы индивидуального течения болезни и выбирать соответствующие методы лечения.
Современная медицинская генетика вооружила клиницистов методами ранней, досимптомной (доклинической) и даже пренатальной диагностики наследственных болезней. Интенсивно развиваются и в некоторых местах уже применяются методы преимплантационной (до имплантации зародыша) диагностики.
ОСНОВНЫЕ ПОЛОЖЕНИЯ КЛИНИЧЕСКОЙ ГЕНЕТИКИ
МЕНДЕЛИЗМ
Основоположником генетики как науки является австрийский естествоиспытатель
Иоганн Грегор Мендель (1822-1884). Он впервые изложил методы генетического анализа и закономерности наследования отдельных признаков организма в работе «Опыты над растительными гибридами». На примере скрещивания различных линий гороха Мендель следил за наследованием индивидуальныхпризнаков в их контрастирующих формах (например, форма семян была круглая или морщинистая, окраска семядолей - желтая или зеленая, окраска цветков -красная или белая и т.д.). Исходными для скрещивания всегда были «чистые линии», т.е. сорта гороха, в течение многих поколений проявляющие лишь одну т форм контрастирующих признаков. Мендель проводил свои опыты в течение 8 лет. Одной из главнейших особенностей метода был точный подсчет результатов каждого опыта (статистический подход), который и позволил установить истинный количественный характер расщепления и сформулировать законы наследственности. Однако работа Менделя долгое время оставалась неизвестной для большинства его современников, и только в 1900г. три ботаника из разных стран-X. де Фриз (Голландия). К. Корренс (Германия) и Э. Фон Чермак (Австрия) независимо друг от друга «псрсоткрыли» менделевские законы. Именно поэтому 1900 г. является официальным годом рождения генетики.
ЗАКОНЫ МЕНДЕЛЯ
Первый закон - единообразие гибридов первого поколения - закон доминирования.
Р: АА х аа
Гаметы: А. А: а. а
Р1: Аа (все - одинаковые)
Результат анализа первого поколения заключается в том. что у всех растений проявлялся только один контрастирующий признак (например, по окраске семядолей все семена первого поколения были желтыми, по окраске цветков - красными, по длине стебля - высокими). Следовательно, у гибридов первого поколения из каждой пары альтернативных признаков развивался только один.
Эффект проявления у гибридов одного из двух контрастир>тощих признаков Мендель назвал доминированием. Таким образом, домннантность - способность вида занимать в сообществе главенствмощее положение и оказывать преобладающее влияние. Соответственно признак - доминантный. Отсутствие фенотипического проявления - рецессивность. Признак подавленный, не проявляющийся в гибридах первого поколения, получил название рецессивного. На характер доминирования направление скрещивания не влияет.
Второй закон - расщепления, или чистоты, гамет.
Р: Аа х Аа
Гаметы: А. а: А. а
Р1: АА: Аа: аА:аа
Во втором поколении расщепление происходит фенотипически (3:1). а геноти-пически - АА(1):Аа(2):аа(1). из которых 2 гомозиготы (АА. аа) и 2 - гетерозиготы (а А. Аа).
Здесь целесообразно ввести некоторые общепринятые современные термины:
- ген-фактор наследственности (участок ДНК с информацией об одном признаке):
— генотип - совокупность генов, хапяктепизмопгах данный опгянизм:
- фенотип - совокупность всех внешних и внутренних признаков и свойств особи, сформированных на базе генотипа в процессе индивидуального развития (онтогенезе);
- геном - совокупность генов, содержащихся в гаплоидном (одинарном) наборе хромосом клетки;
- генофонд - набор генов (аллелей) группы особей популяции, группы популяций или вида, в пределах которых они характеризуются определенной частотой встречаемости;
- генокопия - это заболевания или признаки, внешне копирующие друг друга, но возникающие под воздействием разных генов.
- фенокопия - сходство болезни или признаков, обусловленных только факторами внешней среды с генетически обусловленными заболеваниями (например, для врожденного гипотиреоза установлены 1 фенокопия и 5 геноко-пий).
признак - это фенотшшческое проявление или результат действия гена, фактора окружающей среды или их совместного действия;
аллель - это различные формы (варианты) одного и того же гена, расположенные в одинаковых участках (локусах) гомологичных (парных) хромосом;
- гомозигота - чистолинейные организмы, содержащие две идентичные аллели (АА или аа);
гетерозигота - гибриды, клетки которых имеют две различные аллели (Аа).
Третий закон - закон независимого распределения (комбинирования) неаллельных генов.
Р: ААВВ х аавв
Гаметы: АВ; ав
Р1. АаВв
При дигибридном расхождении гены распределяются по каждому признаку, независимо от других пар признаков. Расщепление по фенотипу во втором поколении при дигибридном скрещивании при условии полного доминирования по двум генам происходит не на 2. а на 4 фенотипических класса в соотношении: 9ААВВ: ЗААвв: ЗВВаа: 1 аавв.
При полигибридном скрещивании расщепление определяется разложением бинома (3+1)". где п - число признаков (неаллельных генов), по которым различают скрещиваемые особи.
Взаимодействие генов
Зачастую признаки формируются при участии нескольких генов, взаимодействие между которыми будет отражаться на проявлении фенотипа.
/- А) взаимодействие аллельных генов:
- доминирование - один аллельный ген полностью скрывает действие другого (примерами доминантных признаков служат: белый локон, брахидакти-лия. габсбургская губа, хондродистрофия: примерами подавленных рецессивных
признаков являются альбинигм. алкаптонурия);
сверхдоминирование - это когда у доминантного гена в гетерозиготном состоянии, иногда отмечается более сильное проявление, чем в гомозиготном;
неполное доминирование-когда гибриды первого поколения проявляют признаки, промежуточные между двумя родительскими формами (промежуточное проявление признака отмечено у растения «ночная красавица», у которого, помимо красного или белого цвета лепестков (соответственно действие доминантного или рецессивного генов), встречаются лепестки розового (промежуточного) цвета:
кодоминирование - если аллельные гены в одинаковой мере активны (кодоминантные) гены, (например, при серповидноклеточной анемии у гетеро-зиготы имеется один нормальный и один дефектный гены, соответственно контролирующие синтез нормального и дефектного гемоглобина; ни один из этих генов не доминирует над другим: у таких индивидов симптоматики заболевания почти нет. или она проявляется в очень легкой форме и лишь в условиях кислородной недостаточности):
множественные аллели - многие гены у разных организмов существуют более чем в двух аллельных формах: это происходит в результате многократного мутирования одного и того же локуса в хромосоме, кроме доминантного и рецессивного имеются промежуточные аллели ( правильнее, различные варианты сочетания двух аллелей.незначительно отличающихся друг от друга содержанием информации об одном и том же признаке), при этом по отношению к доминантному они ведут себя как рецессивные, а по отношению к рецессивному - как доминантные, примером является окраска шкур кроликов, оттенки цвета глаз, форма цветка львиного зева (А > ал > а1' > а). Локусы. для которых характерны множественные варианты существования двух аллелей, названы полиморфными генными локусами. В) взаимодействие неаллельных генов:
Гены, расположенные в разных локусах. как на одной, так и разных хромосомах, называются неаллельными.
эпистаз - подавление одного гена другим: гены оказывающие доминантный эффект, называются эпистатическими генами или генами-супрессора-ми: гены, усиливающие доминантное действие, называются генами-интенсифи-каторами; подавляемые гены - гипостатические (встречается при окраске оперения у домашних кур):
комплементарность - это такое состояние, когда при взаимодействии двух неаллельных доминантных генов (взаимодополняющие гены) возникает новый признак, нередко патологический: примером комплементарности для человека являются патологические фенотипы при ретинобластоме или нефроб-ластоме:
полимерия - различные гены могут оказывать действие на один и тот же признак (однозначные, полимерные гены, полигены): при этом степень проявления признака зависит от числа доминантных аллелей полигенов, такие
признаки называют количественными: примером является пигментация кожи, рост. вес.
С) взаимодействие гена и генотипа:
- эффект положения - открыт в 1925г. А. Стертевантом. это изменение действия доминантного гена (как правило его ослабление) при перемещении гена в геноме в другое место (перемещение гена в другое место наблюдается в случае разрыва хромосомы, когда исходно доминантные гены (до момента разрыва), расположенные вблизи точки разрыва), в большей степени утрачивают свой доминантный эффект по сравнению с генами расположенными дальше от точки разрыва.
Показателями зависимости функционирования гена от генотипа служат экспрессивность и пенетрантность. понятия введены в 1926 г. О. Фогтом и Н. В. Тимофеевым-Рессовским:
- экспрессивность - степень выраженности признака в фенотипе (один и тот же признак может варьироваться, например, различная выраженность ринита у разных больных с ОРВИ):
- пенетрантность - это вероятность проявления признака у разных лиц. имеющих ген. контролирующий этот признак («пробиваемость» гена): пенетрантность измеряется в долях лиц (процентах) - это коэффициент пенетрант-ности:
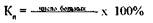
чп<ло гетерсчигот (нос1ггелеп пзтолспмеского гена.)
Пенетрантность может быть полной или неполной, например, при хорее Гентингтона равна 100%. при ОРВИ проявление ринита в 66.6% случаях Основными принципами клинической генетики являются плейотропизм. клинический полиморфизм, генетическая гетерогенность.
- плейотропия (плейотропное действие гена) - зависимость нескольких признаков от одного гена, например, при синдроме Марфана - арахнодактилия. подвывих хрусталика, долихостеномелия обусловлены действием одного гена.
Выделяют первичную и вторичную плейотропию. Первичная - связана с биохимическими механизмами действия мутантного белка или фермента (продукты мутантных аллелей). Например, мутантные аллели различных генов, юнтролирующие синтез коллагена и фибриллина. приводят к нарушению свойств волокон соединительной ткани при синдроме Элерса-Данло. с. Марфана. что проявляется множественными изменениями в фенотипе: деформации скелета, гиперрастяжимость кожи, гиперподвижность суставов и др. Вторичная - обусловлена осложнениями первичных патологических процессов. Например, при талассемии утолщение костей черепа, гепатолиенальный синдром являются результатом вторичного процесса, возникающего в связи с усилением кроветворения, гемосидерозом паренхиматозных органов.
- клинический полиморфизм - это явление вариабельности фенотипа
идентичных по происхождению генов, различия в проявлении одного и того же заболевания, наблюдаемые у разных лиц. имеющих данную патологию, от непроявляемости патологического признака до полной клинической картины. Причинами полиморфизма могут быть: генетический фон. возраст, влияние пола, генетическая гетерогенность, материнские факторы, характер повреждения гена, экзогенные и экологические факторы, эндогенная комплементация и др.
- генетическая гетерогенность - это явление существования различных генов при сходных фенотипах, обусловленное взаимодействием как между отдельными генами, так и между отдельным геном и генотипом. Например, схожая клиническая картина с. Марфана и гомоцистинурии.
НАСЛЕДОВАНИЕ ГЕНОВ И ПРИЗНАКОВ
Наследование - процесс передачи наследственной информации от одного поколения к другому. Варианты и типы наследования базируются на законах наследственности и основных положениях хромосомной теории, зависят от количества наследуемых генов, их природы (доминантнсть или рецессивность) и локализации в хромосомах (на аутосомах или гоносомах) Выделяют 3 варианта наследования генов и признаков. Первые два варианта относятся к классическому, третий - к нетрадиционному наследованию.
Вариант моногенного или простого менделевского наследования Это наследование одного гена или одной пары признаков, оба родителя вносят одинаковый вклад в геном потомка. В рамках этого варианта выделяют следующие типы наследования:
- аутосомно-доминантный. аутосомно-рецессивный:
- Х-сцепленный доминантный; Х-сцепленный рецессивный:
- У-сцепленный.
Критерии моногенного наследования.
Аупюсомно-домииантиый тип:
- доминантные гены, детерминирующие развитие заболвание в гомози-готном состоянии, как правило, легальны.
- гетерозиготы по мутантному ген)- поражены заболеванием:
- заболевание регулярно передается из поколения в поколение без пропусков, т.е. прослеживается по вертикали в родословной, кроме случаев мутации ее поуо (новая мутация);
каждый больной член семьи обычно имеет больного родителя;
оба пола поражаются с одинаковой частотой;
у здоровых детей больного родителя собственные дети и внуки здоровы:
риск рождения больного ребенка, если болен один из родителей, составляет 50%:
к заболеваниям с таким типом наследования относятся хорея Гентингго-
на. с. Марфана. с. акроцефалосиндактилии. нейрофиброматоз I типа и др. Аутосомно-рецессивный тип:
рецессивный ген проявляет свое действие только в гомозиготном состоянии, в гетерозиготном состоянии может существовать во многих поколениях, никак не проявляясь фенотипически;
родители пробанда (лицо, обратившееся за консультацией) здоровы, аналогичное заболевание может обнаружиться у родных, двоюродных и троюродных сибсов (братья и сестры) пробанда. т. е. заболевание прослеживается в родословной по горизонтали (в одном поколении).
если в брак вступают больной с рецессивным заболеванием (аа) и человек с нормальным гомозиготным генотипом (АА). все их дети будут фенотипически здоровыми гетерозиготами:
если в брак вступают больной (аа) и гетерозигота (Аа). в среднем. 50% детей окажутся больными, а 50% - здоровыми гетерозиготами;
- если в брак вступают два здоровых гетерозиготы (Аа). то риск рождения больного ребенка в такой семье равен 25%
в случае кровно-родственных браков между родителями пробанда наблюдается увеличение числа больных в родословной;
мужчины и женщины поражаются аутосомно-рецессивным заболеванием одинаково часто;
примером заболеваний с таким типом наследования являются: гепато-церебральная дистрофия, многие наследственные болезни обмена (ФКУ. гисти-динемия. гомоцистинурия. алкаптонурия. галактоземия). альбинизм, муковис-цидоз и др.).
Наследование заболеваний, сцепленных с полом, определяется тем. что мутантный ген расположен в Х-или У-хромосоме. Известно, что у женщин имеется две половые Х-хромосомы. а у мужчины-одна Х-и одна У-хромосо-ма. У человека в Х-хромосоме локализовано более 200 генов, которые могут быть рецессивными или доминантными. У женщины мутантный ген может находиться в обеих Х-хромосомах или только в одной из них; в первом случае она гомозиготна. во втором гетерозиготна. Мужчины, являясь гемизиготными (имеют только одну хромосому X). передают ее только дочерям и никогда сыновьям. Любой ген, как доминантный, так и рецессивный, локализованный в X-хромосоме. обязательно будет проявляться. В этом главная особенность X-сцепленного наследования.
Х-сцепленный доминантный тип:
- если в Х-хромосоме локализуется доминантный ген. то такой тип наследования Х-сцепленный доминантный;
- заболевание прослеживается в каждом поколении;
болеют как мужчины, так и женщины, однако больных женщин в два раза больше, чем больных мужчин;
если болен отец, то все дочери его будут больными, а все сыновья здоровыми;
если больна мать, то вероятность рождения больного ребенка равна 50%;
больными дети будут, если болен один из родителей по Х-сцепленному доминантному типу наследуются фосфат-диабет, коричневая окраска эмали зубов, с Ретта и др. Х-сцеплеиный рецессивный тип:
- при локализации в Х-хромосоме рецессивного гена: болеют преимущественно лица мужского пола:
- от здоровых родителей могут родиться больные дети (если мать гетерозиготна по мутантному гену):
- признак всегда передается через гетерозиготных матерей, которые фенотипически здоровы:
пораженные мужчины никогда не передают заболевание своим сыновьям:
все дочери пораженных мужчин являются гетерозиготными носительницами:
женщины-носительницы передают заболевание половине своих сыновей (50%). все дочери матерей-носительниц будут здоровы, но половина из них также окажется носительницами:
если в брак вступают здоровый мужчина и женщина-носительница патологического гена, то вероятность рождения больного мальчика составит 50% от всех мальчиков и 25% от всех детей:
вероятность рождения больных девочек очень низка и возможна лишь в том случае, если отец болен, а мать гетерозиготна по мутантному гену, среди девочек половина блдут больны, а другая половина будет нести дефектный ген;
таким образом, наследуется гемофилия (больные страдают повышенной кровоточивостью вследствии недостаточного содержания в крови факторов свертывания крови): болезнь широко распространена в королевских семьях Европы, это связано с заключением близкородственных браков, в результате возникшие мутации сохранялись внутри семьи: королева Великобритании Виктория была носительницей гена гемофилии, и ее сын Леопольд родился гемо-филиком, через своих дочерей и внуков она передала ген заболевания Вольдемару и Генриху Прусским. Фридриху Гессенскому, царю Алексею Романову. Рупрехту Тех-Атлонскому. двум батгенбергским и двум испанским принцам:
кроме гемофилии так наследуются миопатия Дюшенна. некоторые формы дальтонизма и др.
У-сцепленный тип:
в У-хромосоме у мужчин локализовано немного генов: гены передаются только сыновьям и никогда дочерям (голандричсское наследование);
- с У-хромосомой наследуются такие признаки, как гипертрихоз (наличие волос по краю ушных раковин), кожные перепонки между пальцами, развитие семенников, сперматогенез (фактор азоспермии), интенсивность роста тела, конечностей и зубов.
Вариант полигенного наследования
В основе лежит аддитивная полигенная система наследования с взаимодействием 2 и более неаллельных генов и множества факторов внешней среды, не имеющих явного индивидуального эффекта. Аддитивность - суммирование, накопление факторов риска: гены, входящие в состав предрасположенности не являются сами по себе патологическими, а проявление признака будет зависеть от их совместного действия. Иногда полигенное наследование называют мультифакториальным. Критерии полигенной гипотезы были впервые обобщены К.Картером в 1969 г. и дополнены Ф.Фогелем и А.Мотульски в 1989 г.
Критерии полигенного наследования:
- сегрегация признака (заболевания) в семьях (семейных родословных):
- различие популяционных и семейных частот заболеваний; при МФЗ-если заболевание в популяции встречается часто, то семейное накопление относительно меньше, а при редких заболеваниях оно будет выражено чаще. Например, при шизофрении популяционная частота составляет 1%. а семейная - 10% (у родственников 1 степени родства), т.е. десятикратное накопление:
- зависимость риска заболевания от числа больных в семье: чем больше больных родственников, тем выше риск, если оба родителя пробанда здоровы, риск-5-10%. если болен один из родителей - 10-20%. если больны оба. то риск-до 40%;
- зависимость риска от тяжести течения болезни: чем тяжелее течение болезни, тем больше вероятность заболевания у родственников:
- роль полового дисморфизма: если пробанд относится к редко поражаемому полу, то семейное накопление будет выше, и наоборот, если пробанд относится к часто поражаемому полу, то меньше (данный критерий называется эффектом Картера):
- зависимость от наследуемости заболевания: чем выше, тем больше риск (чем больше генов обуславливают заболевание):
- близнецовый критерий: если конкордантность (сходство) монозиготных близнецов по какому-то признаку в 4раза выше конкордантности по этому признаку у дизиготных. то это характерно для полигенного наследования:
если доля больных сибсов в семьях с одним больным родителем выше в 2.5 раза, чем доля больных сибсов в семьях с двумя здоровыми родителями, то предпочтительно полигенное наследование, если меньше 2.5. то нет.
Варианты нетрадиционного наследования
В данном случае речь идет о неканоническом наследовании, которое нельзя строго отнести к моногенному или полигенному варианту, родители вносят неодинаковый вклад в геном потомства. К данному7 варианту наследования относятся:
митохондриальные болезни.
пероксисомные болезни.
болезни геномного импринтинга.
болезни экспансии тринуклеотидных повторов.
- приемные болезни.
- гонадный мозаицичм.
- мейотический драйф.
- инактивация Х-хромосомы. Митохондриальная наследственность:
- передача наследственных признаков осуществляется исключительно по материнской линии (через цитоплазматическую ДНК митохондрий); больны и девочки, и мальчики: больные отцы не передают болезни ни дочерям, ни сыновьям:
- такой тип наследования характерен для болезни Кернса-Сейра. с.Пирсона, атрофии Лебера. МЕЬА8-синдрома. МЕККР-синдрома и др. Гонадный мочаицигн:
- характерно наличие мутации йе поуо в части клеток, предшественников половых клеток, определяющее более высокую вероятность ее унаследования потомством с частотой, заметно превосходящей вероятность мутационного события;
чаще (в 90%) мутации в половых клетках женщины:
- примером являются: несовершенный остеогенез II типа, нейрофибро-матоз 1 типа. с. Дауна (нетранслокационная форма) и др. Мейотический драйф:
- отмечается искажение нормального расхождения хромосом в мейозе. определяющее неслучайную передачу- потомству хромосом и локусов. в них расположенных:
- например, передача потомству генов некоторых моногенных заболеваний: муковисцидоза. миотонической дистрофии. Прионные болезни:
- к ним относятся нейродегенеративные заболевания с поздним проявлением, возникающие в результате действия инфекционного белкового агента, лишенного нуклеиновых кислот:
примером служат такие болезни: б. Крейтцфельда-Якоба. семейная фатальная бессоница. трансмиссионная губчатая энцефалопатия («коровье бешенство»);
- заболевания возникают в результате накопления аномального белка в мозге при мутации гена РКМР. этот белок становится инфекционным агентом (прион - рго1ет 1шесиоп$ а§ет). проявляются атаксией, дегенерацией нейронов, характеризуются медленным течением, поздним проявлением.
Геномный импринтинг (от слова «запечатлеть, отпечаток»):
- эпигенетический процесс, «маркирующий» локусы хромосом одного из родителей, происходит выключение экспрессии генов в них расположенных, т.е. в соответствующем участке генома-моноаллельная. а не биаллельная экспрессия генов (если импринтирован материнский ген. то экспрессия только отцовского):
- неэквивалентный вклад родителей в геном потомства обуславливает отклонение от менделевских законов, меняются фенотипические проявления конкретного гена:
- известно около 30 генов (опухоль Вильмса. с. Прадера-Вилли. с Ан-гельмана):
геномный импринтинг может затрагивать целую хромосому (одноро-дительские диссомии. например при пузырном заносе) и даже геном. Экспансия числа тринуклеотидных повторов:
резкое возрастание числа копий тринуклеотидных повторов в последующих поколениях родословной, сопровождающееся возникновением заболевания при превышении некоторого порогового числа (динамическая мутация);
это постзиготическое событие, происходит на ранних этапах эмбриогенеза;
отмечается также феномен антиципации, т.е. раннее проявление и возрастание тяжести симптомов заболевания в каждом из последующих поколений родословной Данные феномены выявлены при синдроме Мартина-Белла. который обусловлен наличием мутантного гена (РМК1) в сегменте 27 на X-хромосоме. где имеются микросателлитные последовательности ДНК. копии ССС триплета. В норме количество копий от 2 до 54. а в результате мутации происходит увеличение их числа (экспансия) от 54 до 200 - это премутация. не сопровождается клиническими проявлениями, свыше 200 повторов - проявляется на клиническом уровне (полная мутация) Переход от премутации к мутации получил название динамической мутации. Такой переход происходит при передаче гена от матери, причем экспансия зависит от пола ребенка и значительно увеличена при передаче от матери к сыну. Синдром Мартина-Белл широко распространен в популяции - 03-1:1000 у мужчин и 02-06:1000 у женщин. Клинически проявляется: умеренной или глубокой умственной отсталостью, аутизмом, мышечной гипотонией, гиперактивностью, отмечаются большие оттопыренные ушные раковины, прямоугольное лицо, высокий лоб. периор-битальная гиперпигментация, тонкий длинный нос. высокое небо, широкие кисти и стопы, массивный подбородок, макроорхизм. гиперпигментация мошонки, ожирение, гинекомастия. гипоспадия. гиперподвижность суставов, сколиоз, пролапс митрального клапана. Имеются 2 стадии синдрома. 1; премутация, когда нет клинических проявлений; и 2) полная мутация при прохождении через женский мейоз. когда будет клиническая картина. У 80% мужчин имеются признаки синдрома, остальные 20%--здоровы, но при передаче мутации своим детям у них могут быть больные внуки, т.е это мужчины - трансмиттеры (передатчики неэкспрессируемого гена, который становится экспрессируемым при передаче последующим поколениям в родословной).
Феномен экспансии тринуклеотидных повторов описан также при хорее Гентингтона. атаксии Фридрейха. болезне Маччадо-Джозефа, миотонической дистрофии.
ОСНОВЫ МОЛЕКУЛЯРНОЙ ГЕНЕТИКИ
Организация генетического (наследственного материала) осуществляется на следующих уровнях: молекулярном
- хромосомном
- популяционном.
Носителем наследственной информации в живых организмах является молекула ДНК В 1953 г. Дж. Уотсон и Ф. Крик предложили модель структуры ДНК, которая многократно проверялась и признана правильной.
Строение ДНК:
молекула ДНК-это природный полимер, который состоит из двух закрученных вокруг общей оси в правую спираль, полинук-леотидных нитей (цепей). Основная структурная единица одной цепи - нуклеотид (мономер) Он представлен соединенными ко-валентными связями дезоксирибозой, азотистым основанием и фосфатной группой. Последовательность нуклеотидов - это первичная структура ДНК В составе ДНК 4 азотистых основания: 2 пуриновых-аденин (А), гуанин (О) и 2 пиримидиновых - цитозин (С), тимин (Т).
Связь нуклеотидов внутри каждой линейной цепи ДНК обеспечивается чередованием сахара - дезоксирибозы и остатков фосфорной кислоты. Азотистое основание ковалентно соединено с первым атомом углерода дезоксирибозы и формирует структуру, называемую нуклеозидом. Фосфатные группы соединяют соседние нуклеозиды в полимерную цепочку. Обе линейные цепи закручены между собой в правую спираль таким образом, что внутри спирали расположены сахаро -фосфатные группы. Спираль - это вторичная структура ДНК. Каждая цепь имеет два конца:Начало цепи обозначается символом 5'-конец, конец цепи -3'-конец. Цепи ДНК имеют противоположную направленность (антипараллельны). Это позволяет молекуле ДНК сохранять свое постоянство при повреждении каких-то воздействий на нуклеоти-
ды. Вторая цепь выступает в качестве эталона информации. Сцепление между цепями осуществляется водородными связями между аденином (А) и тимином (Т) и между гуанином (г) и цитозином (Ц). Такие специфичные пары азотистых оснований называются комплементарными, а сам принцип построения -комплементарностью пар оснований. Это означает что, когда А и Т противоположных цепей расположены друг против друга, между ними образуются 2 водородные связи, тем самым создается АТ-пара, если это будут Г и Ц, то образуется ГЦ-пара, между которыми 3 водородных связи. Спонтанный процесс образования пар азотистых оснований называется гибридизацией ДНК.
Принято обозначать простую полинуклеотидную структуру как последовательность букв, представленных соответствующими основаниями нуклеотидов Фрагмент двухцепочечной молекулы ДНК с противоположной направленностью цепей можно записать следующим образом:
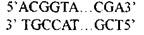
Нуклеиновые кислоты-это очень длинные полимерные цепочки. Интактные молекулы ДНК содержат в зависимости от вида организмов от нескольких тысяч до многих миллионов нуклеотидов. Возможное чис-
 Рис. Схема строения ДНК
Рис. Схема строения ДНК
ло различных последовательностей пар оснований в молекуле ДНК практически бесконечно и способно кодировать колоссальное количество информации.
Строение РНК
РНК - это полимер, состоящий из мономеров, которыми являются нуклео-тиды. Отличиями РНК являются:
- одноцепочечная молекула.
- вместо дезоксирибозы содержится сахар - рибоза.
- вместо тимина - урацил. который будет комплементарен аденину.
- имеется самая разнообразная вторичная конфигурация (конформация).
- коротко живущие молекулы, после завершения своей функции исчезают, постоянно пополняются.
Четыре разновидности молекул РНК:
рибосомальная (рРНК) составляет 80% от общего числа, являются конечным продуктом гена, входящего в состав рибосом (органеллы клетки, на которые синтезируется белок):
транспортные (тРНК) составляет 15%. транспортируют (переносят) АМК к рибосомам. к месту синтеза белка на основе кластерных генов, являющихся конечным продуктом этих генов:
матричная (мРНК-мессенжер или посланник; в отечественной литте-ратуре называется информационной РНК), составляют 5%. несут информацию о структуре белка, конечный продукт - белок:
ядерные (яРНК) - 1%. небольшие молекулы, которые участвуют в созревании мРНК в процессе сплайсинга.
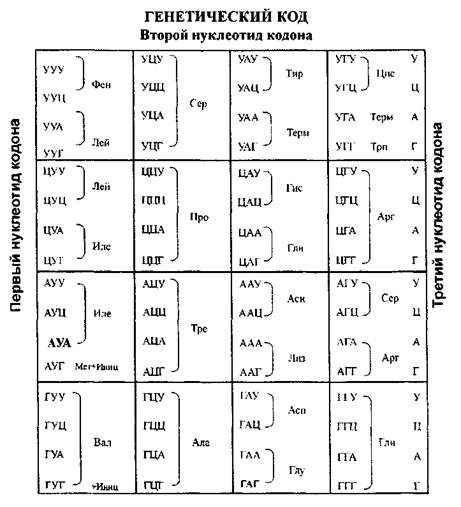
Генетический код
Информация о первичной структуре полипептидов (последовательности аминокислот) записана в ДНК или РНК в виде трехбуквенного кода, составленного из первых букв назвнаий четырех азотистых оснований. Последовательность из трех азотистых оснований называется триплетом или кодоном. Соответствие кодона той или иной аминокислоте составляет сущность генетического кода. Из 64 возможных триплетов 61 кодирует 20 аминокислот, обнаруженных в составе клеточных белков, а 3 кодона являются стоп-сигналами: UAA, UAG, UGA, они прекращают синтез полипсптидной цепи на рибосомах Триплет AUG ( помимо кодирования аминокислоты-метионина) является инициаторным кодоном. с него начинается синтез полипептидной цепи.
Свойства генетического кода
1) триплетность - каждая аминокислота кодируется сочетанием трех нуклеотидов.
2) однозначность - каждому данному кодону соответствует только одна определенная аминокислота.
3) вырожденность генетического кода-одна аминокислота может кодироваться несколькими вариантами триплетов (только две АМК имеют по одному кодону: метионин (AUG) и триптофан (UGG): остальные имеют 2 и более кодонов.
4) неперекрываемость - каждый нуклеотид в мРНК входит в состав только 1 кодона. 5) линейность - триплеты в мРНК прочитываются последовательно в направлении от 5'-конца к 3 "-концу.
6) универсальность - генетический код человека не отличается от кода других эукариотических организмов.
В пределах одного гена, который кодирует полипептид. участок молекулы ДНК подразделяется на функционально различные единицы. Отличительная черта строения многих генов эукариот - прерывистость структуры смысловой части. Смысловые участки, несущие информацию о последовтельности аминокислот в белке -•жзоны, чередуются с участками некодирующих последовательностей -нитронами. В начале каждого гена находятся участки, которые обеспечивают регуляцию работы гена, в частности, способствуют правильной установке рамки считывания нмслеотидов.
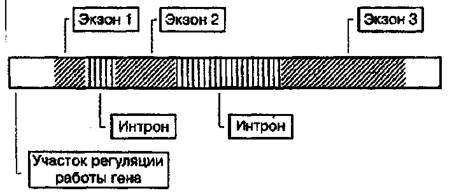
Фундаментальные генетические процессы в клетке Функционирование и сохранность ДНК в ряду поколений обеспечивается работой многочисленных ферментов, которые осуществляют основные генетические процессы в клетке:
репликация ДНК (самоудвоение генетического материала, передача поровну наследственной информации дочерним клеткам);
транскрипция (переписывание или считывание генетической информации, путем синтеза молекулы РНК на основе молекулы ДНК);
- процессинг (созревание молекулы РНК. зрелая матричная РНК содержит необходимую информацию о белке):
- трансляция (процесс синтеза белка на основе зрелой мРНК).
Весь процесс передачи генетической информации от ДНК с помощью различных типов РНК к полипептидам (белкам и ферментам) получила название экспрессия генов. Это однонаправленный поток информации:

Репликация ДНК обеспечивает воспризведение наследственной информации в ряд>' поколений, происходит перед каждым делением клетки. Под действием ряда ферментов в молекуле ДНК разрываются водородные связи, двойная спираль раскручивается и на каждой из разошедшихся цепей как на матрице строится комплементарная ей цепь. Между матричной и материнской цепями вновь образуются водородные связи. Происходит удвоение генетического материала, распределение его в дочерние клетки обеспечивается механизмами митоза (деление соматических клеток) и мейоза (деление половых клеток). Такая репликация называется полуконсервативной, так как одна цепочка сохраняется материнская, а вторая - дочерняя, вновь синтезированная.
В ходе транскрипции РНК-полимераза "узнает" в ДНК промотор (место посадки РНК-полимеразы участок начала транскрипции).перемещаясь вдоль ДНК, последовтельно расплетает двойную спираль ДНК. Одна из цепей ДНК. начиная с промотора, копируется и в ре'5улътатс образуется комплементарная ей мРНК. По мере продвижения РНК-полимеразы. растущая цепь РНК отходит от матрицы, а двойная спираль ДНК восстанавливается позади фермента. Достигнув конца копируемого участка. мРНК отделяется от матрицы. Далее происходит процесс созревания мРНК (процессинг). В начале созревания вырезаются интро-ны. а в конце оставшиеся в ней экзоны. соединяются между' собой (сплайсинг). Усиливают транскрипцию - ънхансеры. а ослабляют - сайленсеры.
В ходе трансляции информация "переводится" в определенную последовательность аминокислот в синтезируемых белках. Этот процесс осуществляется сложным макромолекулярным комплексом, который включает рибосомы. мРНК. тРНК. ферменты, белковые факторы инициации (начало), элонгации (наращивание полипептида). терминации (окончание) трансляции и др.
Ген - это функционально неделимая единица наследственного материала, участок молекулы ДНК. кодирующий первичную структуру полипептида. Длина гена, согласно модели общей структуры гена. - это участок ДНК. имеющий слева 5 "-конец, а справо 3"-конец, между которыми расположены экзоны и интроны
Размеры генов вариабельны. Один из самых коротких генов - ген бета-глобина содержит 1100 пар нуклеотидов (пн), а примером протяженного гена служит ген дистрофика, имеющий 2.6 млн. пн. Общее число генов человека составляет около 70 тысяч, у кишечной палочки -4 тыс.. у дрожжей - 7 тыс.. дрозофилы -15-20 тыс.
Наряду с кодирующими последовательностями немалая доля принадлежит некодирующим, никогда не экпсрессирующимся (не транскрибирующимся) последовательностям - псевдогенам или ложным генам.
Геном человека
Длительное время геномом называли гаплоидный набор хромосом. В настоящее время он означает полный состав ДНК клетки, т.е. совокупность всех генов и межгенных участков. Геном - это полный набор инструкций для формирования и функционирования индивида.
Принципы построения геномов и их структурно-функциональную организацию изучает геномика. которая проводит секвенирование. картирование и идентификацию функций генов и внегенных элементов.
Характеристика генома человека
Общее количество ДНК в соматической клетке составляет 6.4х 10' пар нуклео-тидов. следовательно, гаплоидный набор состоит из 3.2х 109пар нуклеотидов. которые распределены по 25 хромосомам: 22 аутосомы. Х-хромосома. У-хромосома (количество ДНК. локализованное в хромосомах составляет 95%) и «митохондри-альная хромосома» (5%). небольшое количество составляют отдельные кольцевые молекулы ДНК в ядре и цитоплазме.
Внехромосомные и кольцевые молекулы ДНК обнаруживаются в цитоплазме и в ядре. Они изучены еще недостаточно. В строгом смысле они являются не составными элементами генома. а его продуктом Их размер колеблется от 150 до 20 000 пар нуклеотидов.
Хромосомная ДНК делится на две группы: с уникальной последовательностью нуклеотидов и повторяющимися последовательностями. В клетке примерно 50% ДНК с уникальными последовательностями и 50% - с повторяющимися Участки с повторяющимися последовательностями различаются по длине каждого повтора и числу повторов (их называют тандемными):
микросателлитные. состоят из 2-8 пар нуклеотидов: минисателлитные. от 10 до 100000 пар нуклеотидов: повторяющиеся последовательности (некодирующая ДНК) - это районы между генами (спейсеры) или межгенные промежутки.
Главной формой генетического полиморфизма является однонуклеотндный полиморфизм (ОНП). Под этим термином понимают варианты последовательностей ДНК у разных людей с вовлечением одной пары нукледотидов. ОНП - наиболее общий источник вариаций между людьми. Эти вариации встречаются на протяжении всей ДНК (в экзонах. интронах. мсжгенных промежутках, повторах) и отражают прошлые мутации. Установлено, что однооуклеотидные различия обнаруживаются на протяжении 1 000-2 000 нуклеотидной длины. Это означает, что на всю длину генома должно быть 1.6-3.2 млн ОНП. К 2001 г. идентифицировано и картировано 1.42 млн ОНП. Расчеты показывают, что два человека на 99.9% идентичны по нуклеодитным последовательностям, т.е. только 0.1 % различий по одному нуклиотиду создает такие огромные индивидуальные фенотипические вариации, которые легко видеть в любой группе индивидов.
Митохондриальный геном представлен кольцевой двухцепочечной молекулой, содержащей 16 569 пн. кодирует две рибосомные РНК (125 и 165). 22 транспортные РНК и 13 полипептидов. Митохондриальный геном отличается отядерного генома несколькими признаками:
мтДНК наследуется по материнскому типу, доля отцовской мтДНК в зиготе составляет от 0 до 4 митохондрий, а материнских - 2 500. репликация отцовских митохондрий после оплодотворения блокируется:
комбинативная изменчивость мтДНК (мейоз) отсутствует, нуклсотидная последовательность меняется в поколениях только за счет мутаций:
гистоны и негистоновые белки. Гистоны характерны только для эукариотически.х клеток, они осуществляют первые этапы упаковки ДНК. Их взаимодействие с ДНК происходит за счет ионных связей. Это глобулярные белки, представленные 5-7-ю видами молекул. Наиболее известны следующие классы гистонов: Н1. Н2А. Н2В. НЗ. Н4. Их основные свойства определяются относительно высоким содержанием аминокислот, лизина и аргинина.
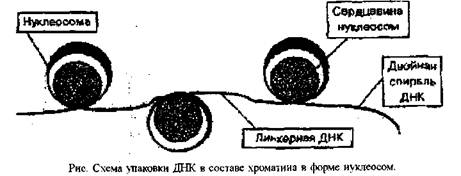
Первый этап упаковки ДНК в хроматине осуществляется с помощью муклеосам. Молекула ДНК накручивается на поверхность множества белковых частиц, каждый раз делая 1.75 оборота вокруг сердцевины. Сердцевина нуклеосомы всегда консервативна и содержит 8 молекул - по 2 молекулы гистонов Н4. НЗ. Н2 А. Н2В. По поверхности сердцевины располагается участок ДНК из 146 нуклеотидных пар. Небольшой участок ДНК между нуклеосомами остается несвязанным с сердцевиной, он называется линкером. Общий вид хроматина, представленного молекулой ДНК. упакованной с помощью нуклеосомных структур, можно сравнить с бусами на нитке. Первый нуклеосомный уровень компактизации укорачивает молекулу ДНК в 6-7 раз.
В следующий этап упаковки нуклеосомная структура хроматина вовлекается с помощью гистонба Н1. который связывается с линкерной частью ДНК и поверхностью нуклеосомы. Возникает упорядоченная структура спирального типа, кото-р>то называют соленоидом. Она повышает компактность ДНК еще в 40 раз. под электронным микроскопом соленоид выявляется в виде фибрилл хроматина. Фибриллы ДНК попарно скручиваясь, образуют хромонемы (греч. пета-струна). которая входит в комплекс более высокого порядка полухроматиды (спирально закручены). Дальнейшая суперспирализация хроматина приводит к образованию пары хроматид, а пара хромата образует хромосому. Хроматиды соединяются между собой в районе первичной перетяжки илицентромеры. Центромеры выполняют в хромосомах очень важные функции, они соединяют две сестринские хроматиды играют важную роль в организации веретена деления (на ней находится кинетохор. к которому присоединяются нити веретена при делении). Внутри центромеры выявляются уникальные последовательности, которые, вероятно, несут информацию о расхождении хромосом к противоположным полюсам клетки. Благодаря первичной перетяжке хромосома делится на плечи, короткое (р) и длинное (С|).
В 'зависимости от размера, положения центромеры и длины плеча выделяют три типа хромосом:
медианный (метацентрический) - центромера расположена в середине хромосомы и оба плеча равны;
субмедианный (субметацентрический) - центромера расположена ближе к одному из концов, плечи хромосомы не равны:
субтерминш!ьный (акроцентрический) - центромера расположена на конце хромомсомы.
Для измерения длины хромосомы был предложен центромерный индекс - доля длины короткого плеча длине всей хромосомы, принятой за 100%. Если центромерный индекс равен 50%. то это метацентрическая хромосома, если меньше 50% - субметацентрическая хромосома, а если центромера расположена близко к концу хромосомы - то акроцентрическая.
Концевая часть хромосомы называется теломерой, в ней находятся повторяющиеся последовательности ДНК - теломерная ДНК. препятствующая укорочению хромосомы при ее репликации.
Хромосома не однородна, выделяют активные районы, которые содержат гены, контролирующие развитие признаков организма; в интерфазных ядрах это слабо-окрашивающиеся нитчатые структуры, хроматин, представлен здесь в активной форме, в нем происходит транскрипция - синтез всех типов молекул РНК Эти районы называют эухроматином. Также выделяют неактивные участки, они отличаются высокой спирализацией. сохраняются на протяжении всего митатического цикла, интенсивно окрашиваются, не содержат уникальных генов, и получили название гетерохраматин. Гетерохроматин делят на структурный (фрагменты прилегающие к области центромеры, а также расположенные на свободных теломе-рах) и факультативный (образуется при спирализации одной из двух негомологичных хромосом, его роль - в компенсации дозы определенных генов).
В некоторых хромосомах выделяют вторичную перетяжку, которая расположена ближе к концу хромосомы и образует ее спутник (сателлит) - на 1. 9. 16.
Хромосомы клетки можно увидеть только во время ее деления - при митозе (форма деления соматических клеток) и мейозе (форма деления половых клеток). их называют метафазные пластинки.
Совокупность морфологических особенностей полного хромосомного набора свойственного соматическим клеткам вида Ното 5ар!еп5 (определяется общим количеством хромосом, их формой и размерами) называется кнрнотипом человека.
Нормальный кариотип человека - 46.ХХ (женщина) и 46.ХУ (мужчина), те диплоидный (двойной) набор хромосом, или 22 пары аутосом (хромосомы тела) и 1 пара гоносом (половые хромосомы). Впервые термин был введен в 1924 г. Г.А. Левитским. У женщин гоносомы представлены двумя гомологичными (идентичными) Х-хромосомами. так как в процессе оогенеза образуется один тип яйцеклеток, содержащих Х-хромосому У мужчин гоносомы представлены одной Х-хро-мосомой и одной У-хромосомой. так как в процессе сперматогенеза образуются
два типа сперматозоидов, имеющих Х-хромосому либо У-хромосому (их соотношение примерно 1:1). Определение пола человека происходит в момент оплодотворения яйцеклетки сперматозоидом. Оно целиком зависит от того, какой это сперматозоид: если с Х-хромосомой. то будет женский организм, если с У-хромосомой. то -мужской организм. Для изучения кариотипа человека используют клетки костного мозга, культуру фибробластов, лейкоциты, периферической крови. Предложены три методических приема для изучения хромосом: культивирование клеток, их обработка гипотоническим раствором и использование для остановки клеточного деления алкалоида - колхицина В 1960г. Р. Мурхед впервые предложил метод получения метафазных пластинок из лимфоцитов крови, культивируемых с фитогемагглютинииом. Этот метод стал основой стандартного метода анализа метафазных хромосом.
Классификация и методы окрашивания хромосом
В г.Денвере (США) в 1960 г. проводится первая международная научная конференция цитогенетиков. на которой предлагается номенклатура хромосом человека в зависимости от их морфологической характеристики, учитывающей размеры, форму и положение центромеры. Согласно денверской классификации все аутосо-мы получили порядковые номера и были разделены на 7 групп:
А (1 -3 пары) - наиболее крупные метацентрические.
В (4.5) - большие субметацентрические.
С (6-12) - средние сл'бметацентрические.
О (13-15) - большие акроцентрические.
Е (16-18) - малые субметацентрические.
С (21.22) - малые акроцентрические.
Внутри этих групп пары хромосом практически не различимы. По принципу морфологического подобия Х-хромосома не отличима от хромосом группы С. а У-хромосома - от хромосом группы С.
Для точной идентификации хромосом разработаны различные методы дифференциального окрашивания Наиболее распространенным методом является
6-окрашивание (Гимза). с помощью его препараты хромосом можно анализировать под обычным световым микроскопом.
0-окрашивание (акрихин) - это флюорссцентное окрашивание, четко выявляет У-хромосому.
Исчерченность хромосом при С- и Р-окрашивании почти всегда совпадает, за исключением прицентромерных районов некоторых хромосом.
Я-окрашивание (геуегз - обратный) выявляет обратную картину по сравнению с (5-окрашиванием. хорошо выявляет теломерные районы.
С-окрашивание (конститутивный гетерохроматин) позволяет выявить сегменты центромерных и околоцентромерных участков всех хромосом, коротких плеч 13.14, 15.21.22 и длинного плеча У-хромосомы. структурный гетерохроматин.
Метод дифференциального окрашивания сестринских хроматид выявляет различия между нормальными и измененными клетками при ряде наследственных заболеваний, между механизмами обменов, позволяет использовать частоту СХО
как тест на мутагенную активность физических факторов и химических соединений, включая лекарственные препараты.
Метод дифференциального окрашивания хроматид с помощью бромдсзоксиу-ридина (БУДР). являющегося аналогом тимина. почволяет идентифицировать различные типы сегментов (блоки) хромосом.
В начале 80-х годов XX столетия широкое распространение получили более совершенные методы дифференциального окрашивания хромосом, позволяющие анализировать мало компактизованные хромосомы на стадиях профазы и прометафазы (профазные и прометафазные методы) и проводить разделение нормального гапло-идного набора хромосом более чем на 2000 структурных элементов (сегментов).
Изменчивость наследственного материала
Модификационная изменчивость не затрагивает генотип особи, не передается при половом размножении и отражает изменение фенотипа под действием… не передается следующим поколениям. - яатяется адаптивной (приспособительной);КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
С генетической точки зрения все болезни в зависимости от относительной значимости наследственных и срсдовых факторов в их развитии можно подразделить на 3 группы:
1. Наследственные (юлети, ~ этиологическим фактором которых являются мутации, практически проявление патологического действия мутации не зависит от среды: последняя может только менять выраженность симптомов болезни и тяжесть ее течения, к заболеваниям этой группы относятся хромосомные болезни, генные болезни.
2. Болезни с наследственной предрасположенностью (мультифакгориальные. многофакторные болезни, болезни взрослой жизни) - болезни развиваются у лиц с определенной генетической характеристикой под влиянием факторов окружающей среды (т.е. суммарное, аддитивное действие множества факторов): к заболеваниям этой группы относятся большинство хронических неинфекционных болезней (сахарный диабет, атеросклероз, экзема, псориаз. ИБО. язвенная болезнь желудка и др.). фармако- и экогенетические болезни, изолированные врожденные пороки развития.
3. Ненаследственные болегни - в происхождении этих заболеваний определяющую роль играет среда, генегические факторы могут влиять только на течение патологических процессов (выздоровление, компенсация нарушенных функций и др.): к этой группе относятся большинство травм, инфекционные болезни, ожоги, обморожение, лучевая болезнь.
Генетическая классификация наследственных болезней
В основу классификации положен этиологический принцип, а именно тип мутации и характер взаимодействия со средой: всю наследственную патологию делят на пять групп:
1. генные болезни (моногенные).
2. хромосомные болезни (ХБ).
3. болезни с наследственной предрасположенностью (МФБ).
4. генетические болезни соматических клеток.
5. болезни генетической несовместимости матери и плода.
Генные болезни обусловлены мутацией 1 гена (генные, точковые мутации), передаются из поколения в поколение в соответствии с законами Менделя (мендели-рующие заболевания). В настоящее время известно около 5000 заболеваний.
Хромосомные болезни вызваны хромосомными и геномными мутациями. Большинство ХБ. обусловленыо анеуплоидиями. не наследуются. Структурные перестройки передаются с дополнительными перекомбинациями, возникающими в мейозе носителя перестройки, последующим поколениям. Насчитывается около 600 хромосомных болезней.
Болезни с наследственной предрасполженностью могут быть моногенными и полигенными; наследственная предрасположенность реализуется с помощью сре-дового фактора. Генетические болезни соматических клеток выделены в отдельную группу наследственной патологии недавно. При злокачественных новообразованиях обнаруживаются специфические хромосомные перестройки в клетках, вызывающих активацию онкогенов (ретинобластома. опухоль Вильямса). Эти изменения в генетическом материале клеток являются этиопатогенетическими для злокачественного роста и поэтому могут быть отнесены к категории генетической патологии. Имеются первые доказательства того, что спорадические случаи ВПР являются результатом мутаций в соматических клетках в критические периоды эмбриогенеза: весьма вероятно, что аутоиммунные процессы, старение могут быть отнесены к этой же категории генетической патологии
Болезни, возникающие при несовместимости матери и плода по антигенам, развиваются в результате иммунной реакции матери на антигены плода. Кровь плода в небольшом количестве попадает в организм беременной. Если плод унаследовал от отца такой аллель (антиген. Аг+). которого нет у матери (Аг-). то организм беременной отвечает иммунной реакцией. Антитела матери, проникая в кровь плода, вызывают у него иммунный конфликт. Наиболее типичное и изученное заболевание этой группы гемолитическая болезнь новорожденных.
Клиническая классификация наследственных болезней делится
1. По органному, системному принципу,
наследственные заболевания нервной системы, болезни опорно-двигательного аппарата, болезни крови и т.д.
2. В зависимости от нарушения обмена веществ. Такая биохимическая классификация как бы объединяет генетический и физиологический подходы. По такому принципу различают наследственные болезни обмена:
углеводов (галактоземия).
липидов (ганглиозиды. муколипидозы).
- аминокислот (фенилктонурия. гистидинемия).
- витаминов (витамин 0-зависимый рахит).
- пуринов и пиримидинов (подагра, с. Леша-Ниихана) и др.
Кроме того, в рамках наследственных болезней обмена отдельно выделяют болезни накопления (тезаурисмозы) Эта группа заболеваний вызвана недостатком лизосомальных ферментов ферментов и проявляется прогрессирующим отложением веществ определенного типа (обычно предшественники реакций) в клетках различных тканей.
Особенности проявления наследственной патологии (основные признаки, по которым можно заподозрить наличие у больного наследственной патологии):
1) Семейный характер заболевания - характерна сегрегация (накопление) симптомов заболевания в семье пробанда. что обусловлено наличием общих генов у членов семьи.
2) Хроническое, прогредиентное. рецидивирующее течение. Для наследственной патологии нетипично улучшение состояния, а значит исчезновение симптомов. Например, хроническая пневмония с бронхоэктазами формируется у детей с
легочной формой муковисцидоза. Хронический процесс развивается в результате постоянного действия мутантного гена.
3) Специфические симптомы наследственных болезней, диагностическое значение микропризнаков Наличие у больного редко встречаемых специфических симптомов или их сочетание дает основание думать о врожденной или наследственной природе заболевания. Например, голубые склеры при несовершенном остеогенезе и других заболеваниях соединительной ткани, долихостеномегалия при с. Марфана. «мышиный запах» при фенилкетонурии и др.
4) Недоразвитие или чрезмерное развитие отдельных частей тела. Связано с на-Р)тлением на ранних стадиях онтогенеза. Воздействие генов на морфологическом уровне проявляется в виде дегенерации, либо усиление пролиферации клеток определенной ткани, либо процесс нормальной дегенерации или пролиферации осуществляется в необычное для них время. Вследствие этого происходит нарушение взаимодействия между зачатками или клеточными слоями в онтогенезе и др. процессы, следствием чего яатяется развитие более или менее специфических симптомов наследственной патологии, в зависимости от времени проявления мутантного гена, а именно недоразвитие или чре'лмерное развитие отдельных частей тела.
5) Множественность (полисистемность) поражения при наследственной патологии. Вовлечение в патологический процесс нескольких систем и органов обусловлено плейотропным эффектом гена.
6) Клинический полиморфизм характерен для всей наследственной патологии, в его основе лежат такие генетические причины, как множественный аллелизм. генокопировние. геномный импритинг и др.
7) Задержка физического и психического развития
8) Снижение продолжительности жизни.
9) Согласовнность времени манифестации и характера нарушения с этапами онтогенеза
10) Резистентность к наиболее распространенным методам терапии.
11)Этническая приуроченность патологических признаков.
ГЕННЫЕ БОЛЕЗНИ (МОНОГЕННЫЕ)
Генныеболезни (ГБ) - это заболевания, подчиняющиеся менделевскому наследованию, обусловленные точковыми мутациями (мутации в одном гене), которые включают: дефекты экзонов. нитронов, фланкирующих областей, приводящие к изменению состава или порядка нуклеотидов в молекуле ДНК. нарушению трансляции генетической информации от ДНК к РНК и от РНК на рибосомы. в результате чего изменяется последовательность аминокислот в полипептидс.
В настоящее время известно около 4860 заболевний этой группы. Генная патология занимает значительное место в современной клинической медицине.
Условно принято считать, что ГБ - часто встречающиеся, если их частота 1:10 тысяч и выше, редие. если частота 1:100 тысяч и менее. Насчитывается около 700-900 нозологических единиц, гены которых картированы на хромосомах.
Генные болезни в зависимости от типа наследования делятся на: аутосомно-дом ина нтные.
- аутосомно-рецессивные.
- Х-сцепленные. доминантные болезни
- Х-сцепленные. рецессивные болезни.
В зависимости от вовлечения органов и систем выделяют болезни нервной, сердечно-сосудистой системы, органов дыхания, соединительной ткани, кожи и т.д.
По этиологии выделяют 2 класса 'заболеваний:
болезни с установленным первичным молекулярным (биохимическим) дефектом, т.е. наличие дефектного гена и нарушение биохимической реакции, в которой участвует белковый продукт нормального гена; около 500 болезней (10%):
- болезни с неустановленным первичным (биохимическим) дефектом, около 90% всех ГБ.
В зависимости от нарушения вида обмена веществ (наследственные болезни обмена) выделяют - болезни аминокислот, углеводов, липидов. минералов и т.д.
Показаниями для диагностики при подозрении на генное заболевание являются:
1) задержка психомоторного и физического развития ребенка на первом году жизни в сочетании со следующими признаками:
- прогрессирующая умственная отсталость.
резистентные к терапии эпилептические припадки, прогредиентная неврологическая симптоматика (парезы, мышечная гипотония и др.).
рвота.дегидратация.желтуха. асцит.диарея.
- нарушения дыхания, летаргия, кома, необычный запах тела и мочи.
- гипотрофия.
гепатомегалия. спленомегалия неясной этиологии.
- метаболический ацидоз, алкалоз, изменение показателей сахара, белка, ацетона в моче, лейкоцитопения. тромбоцитопения. изменение иммунологических показателей:
2) в последующие годы жизни ребенка:
прогредиентное развитие умственной отсталости, неврологической симптоматики после периода кажущегося благополучия.
гипотрофия неясной этиологии.
непереносимость отдельных продуктов питания.проявляющиеся анорек-сией. диареей, задержкой развития.
- изменение лабораторных данных (см. выше), нефролитиаз.
умственная отсталость в сочетании с задержкой физического развития, лицевыми дизморфиями. гидроцефалией. глухотой, изменениями со стороны органов зрения, гепато/спленомегалией. поражением почек, судорогами, алалией. комой, летаргией и др. нарушениями. Характеристика отдельных помологических форм. Фенилкетонурия
Наследственная болезнь обмена аминокислот, характеризуется прогрессирующим слабоумием, обусловлена генетически детерминированным дефектом соответствующих ферментов (ферментопатия). В настоящее время выделяют несколько типов заболевания, которые зависят от мутаций различных генов.
1тип ФКУ: описал Фоллинг в 1934 году, ген картирован на длинном плече 12 хромосомы (12^22-^24.1). отмечается дефицит фермента фенилаланин - 4-гидро-ксилазы. Характерен аутосомно-рецессивный тип наследовния. частота заболевания 1:8 тысяч. Отмечается метаболический блок на уровне перехода фенилалани-на в тирозин. В жидких средах организма (кровь, моча. СМЖ) повышается концентрация ФА. снижается уровень тирозина. Происходит активация побочного пути превращения ФА - переаминирование с альфа-кетоглутаровой кислотой и образование фенилпировиноградной. фенилмолочной. фенилуксусной кислот, оказывающих выраженное токсическое действие на ЦНС. паренхиматозные органы, вызывая необратимые изменения в них.
Угнетается активность ряда других ферментов (трансаминаза. декарбоксила-за). появляются вторичные нарушения обмена аминокислот, тирозина, трштгофа-на. ДОФА. нарушается синтез катехоламинов (ноадреналина. адреналина). Клиника: у новорожденных никаких изменений не отмечается. Первые признаки у ребенка появляются около 2-6 месяцев, после «светлого промежутка», возможен респираторный и нейродистресс синдром, судороги, рвота, беспокойство, экзематозные изменения на коже, специфический запах мочи и пота («мышинный»). задержка моторного, психического развития. Характерен внешний вид больных: светло-русые волосы, слабая пигментация кожи, светло-голубые глаза (частичный альбинизм связан с нарушением синтеза меланина).Ведущими клиническими симптомами являются: прогрессирующая умственная отсталость.эпилептические пароксизмы (симптоматическая эпилепсия).психотические расстройства.
Лабораторная диагностика:
- неонаталъный скрининг ФКУ. на 3-5 день жизни всем доношенным новорожденным берется кровь из пятки (недоношенным - на 7-14 день), наносится на специальную фильтровальную бумагу, высушивается, отправляется в лабораторию скрининговых программ, где с помощью ЙФА определяется концентрация фени-лаланина в крови;
- микробиологический тест Гатри (исследуется рост В.зиЫШз); качественная проба мочи с 10% РеС13:
биохимические методы определения концентрации фенилаланина в крови и моче путем флюорометрического метода, бумажной хроматографии. высоковольтного электрофореза, хроматографии на ионообменных смолах:
ДНК-диагностика ФКУ Лечение:
Для лечения наследственных болезней обмена аминокислот разработаны следующие основные подходы:
- ограничение поступления белка и соответствующих аминокислот с пищей.
- дополнительное введение незаменимых аминокислот.
- использование препаратов, активирующих альтернативные пути метаболизма.
- усиление связывания и выведение накапливающихся продуктов обмена, введение кофакторов энзимных реакций (биоптерин).
- симптоматическая терапия (противосудорожные препараты, ноотропы. витаминотерапия и др.).
интенсивная терапия в острый период (гемофильтрация. перитонеальный диализ).
Для вскармливания детей раннего возраста используются белковые гидролиза-ты с низким содержанием фенилаланина и повышенным тирозина - лофеналак. фенил-фри. кетонил. цимогран. берлофен.
IIтип ФКУ:описал ЗтИЬ. 1974. ген- на 4 хромосоме (4р15.31). отмечается дефицит дигидроптеридинредуктазьк манифестирует на первом году жизни, летальный исход к 2-3 годам. Отсутствует эффект от диетотерапии, в лечении используются препараты Ь-дофы.
Штнп ФКУ:описал КаиГтап в 1978 году, ген не уточнен, частота-1:30 тысяч, отмечается дефицит кофакторов синтеза фенилаланина. Ведущими клиническими симптомами являются: умственная отсталость, спастический тетрапарез. тонико-клонические судороги, микроцефалия.
Синдром Марфана
Клиника: поражаются преимущественно опорно-двигательная, сердечно-сосудистая система и органы зрения. Со стороны скелетной системы: высокий рост, астеническое телосложение.ХРОМОСОМНЫЕ БОЛЕЗНИ
Хромосомные болезни (синдромы) - клинически значимая патология, общая частота в популяции составляет около 1%. она регистрируется у 5-6%… В 'зависимости от того, имеется ли мутация в системе аутосом или в системе… Мутация может возникнуть заново в гаметах здоровых родителей (мутация de novo, спорадические случаи), или родители уже…Синдром Эдвардса - трисомия 18
Клинически отмечается выраженная задержка пренатального развития при полной продолжительности беременности Для синдрома характерны множественные… Дети с синдромом Эдвардса погибают до года.Синдром Патау - трисомия 13
Синдром «крик кошки» - синдром делении короткого плеча 5. Хромосомы
Ответственен за развитие синдрома сегмент 5р 15.1 -15.2. Частота -1:50000. среди детей с задержкой умственного развития - 1:350. Описаны мозаичные… Дети, как правило, рождаются после нормально протекавшей беременности. В… Синдром Вольфа-Хиршхорна (синдром делении короткого плеча 4 хромосомы)Синдром Клайнфельтера
Частота - 1:500 новорожденных мальчиков. Цитогенетические варианты: подавляющее большинство больных имеют кариотип - 47. ХХУ.Синдром трисомии X
Синдром полисемии У Частота 1:1000 новоржденных мальчиков. Кариотип: 47.ХУУ Для пациентов… Показания к цитогенетическому исследованию:ВРОЖДЕННЫЕ АНОМАЛИИ РАЗВИТИЯ
По данным ВОЗ популяционная частота ВПР составляет 1.5-5% (в отдельных странах - 2.7-16.3 %). На 1000 рождений врожденные дефекты развития… Врожденный порок развития (ВПР, большая аномалия развития)- это стойкое… ВПР входят в структуру «врожденных аномалий» (соп§ет1а1 апота1у), или «врожденных дефектов»(ЫнН Ае/ес1), данные…Международная классификация врожденных пороков развития
По частоте:
распространенные (1: 1000 и более новорожденных).
умеренно частые (0.1-0.99 на 1000 новорожденных).
очень редкие (меньше 0.01 на 1000 новорожденных) По тяжести:
летальные (смерть до репродуктивного периода) - 0.6%.
средней тяжести (не угрожает жизни, но требует оперативного вмешательства)-1.9-2,5%.
- малые аномалии развития или информативные морфологические варианты-3,5%.
По проявлению:
изолированные (одиночные).
системные.
- множественные (синдром, ассоциация, случайные комбинация). По этиологии:
моногенные - 6%.
хромосомные - 5%.
- внешнесредовые (тератогены. материнские факторы) - 6%.
- мультифакториальные - 63%. неустановленные причины - 20%.
Дисморфологическая классификация:
Мальформация (таИогтаиоп) - морфологический дефект органа, части органа или большого участка тела в результате внутреннего нарушения процесса развития (генетически детерминированный процесс).
Дизрупция (Швгарйоп) - морфологический дефект органа, части органа или большого участка тела в результате внешнего препятствия или какого-либо воздействия на изначально нормальный процесс развития (тератогенные факторы и нарушение имплантации).
Деформация (йеГогтаиоп) - нарушение формы, вида или положения часта тела, обусловленное механическими воздействиями.
Дисплазия (<1у5р1а51а) - нарушенная организация клеток в ткани (тканях) и ее морфологический результат (процесс и следствие дисгистогенеза).
В зависимости от времени и объекта воздействия вредных факторов ВПР делятся на четыре типа:
гаметопатии - связаны с возникновением %гутаций в половых клетках родителей (генные, хромосомные синдромы);
бластопатии - поражение бластоцисты (зародыш первых 15 дней после оплодотворения, следствие бластопатии - двойниковые пороки, циклопия. сиреноме-лия).
гнбриопатии - возникают в период от 16 дня до конца 8 недели беременности (в этот период происходит активный органогенез и гистогенез, большинство пороков образуется в этот период):
фетопатии - повреждение плода в период от 9 недели до окончания беременности (крипторхизм. тазовая почка, метаболические фетопатии).
Для объяснения этиологических и патогенетических связей между разными типами аномалий развития были предложены следующие понятия, следствие, синдром, ассоциация.
Следствие - это тип множественных аномалий, возникших в результате одной известной или предполагаемой аномалии, либо в результате действия механического фактора. Например, спорадический аномалад Пьера-Робена. при котором первичным пороком является микрогения, а его следствием будут уменьшение ротовой полости, глоссоптоз. расщелина неба.
Синдром - устойчивое сочетание двух и более пороков развития, выявляемых в разных системах организма
Ассоциация - это неслучайное сочетание нескольких аномалий развития у двух или более индивидов, неизвестное как синдром или следствие. К наиболее распространенным и часто встречающимся в практике врача любой специальности относятся следующие ВПР (БАР):
агенезия - полное врожденное отсутствие органа:
аплазия - врожденное отсутствие органа с сохранением его сосудистой ножки:
атрешя и стеною - соответственно полное отсутствие или сужение канала (отверстия):
врожденная гипоплазия - недоразвитие органа, проявляющееся дефицитом относительной массы (отношение абсолютной массы органа к абсолютной массе тела плода или ребенка, выраженное в процентах) или размеров орггана. превышающее отклонение от средних показателей в две сигмы (средняя ошибка) для данного возраста; простая - без нарушения структуры и диспластическая - с нарушением структуры;
- врожденная гипотрофия - уменьшение массы тела плода или новорожденного:
врожденная гипертрофия (гиперплазия) - увеличение относительной массы (размеров) органа за счет увеличения объема (гипертрофия) или количества (гиперплазия) клеток;
- гетероплазия - нарушение дифференцировки отдельных типов тканей, например, клетки плоского эпителия пищевода в дивертикуле Меккеля;
гетеротопия - наличие клеток, тканей или участка органа в другом органе или в тех зонах органа, где в норме их не должно быть;
- эктопия - смещение органа в нетипичное для него место;
дизрафия или арафия - незаращение эмбриональной щели (расщелина губы или неба, позвоночника);
дисхрония - нарушение темпов развития (ускоренное или замедленное);
макросами» (гигантизм) - увеличение длины тела;
неразделение (слияние) - может быть органа или монозиготных близнецов (краниопаги. торокопаги. сросшиеся черепами или грудными клетками близнецы);
- персистирование - сохранение эмбриональных структур, в норме исчезающих к определенному периоду онтогенеза, например, незаращение баталлова протока или овального окна у ребенка в возрасте старше трех месяцев: нарушение лобуляции - дополнительная доля легкого, печени: инверсия - зеркальное расположение органов;
- удвоение органов}
образование водянок - гидроцефалия. гидронефроз
Мониторинг ВПР
ственности к проблемам возможного вредного воздействия факторов окружающей среды на человека и потомство. Во всем мире уже в течение нескольких… Основная практическая задача мониторинговых регистров - получить максимально… анэнцефалия - расщелина небаАкроцефалосиндактилия
Синдром Алера (акроцефалосиндактилия тип I ). Наиболее распространенный, частота 1:100-160 тыс. новорожденных,… довременного сращения затылочной кости. Редш пороки сердца, атрезия заднего прохода, пилоростеноз. атрезия пищевода.…Синдром Дубовица.
БОЛЕЗНИ С НАСЛЕДСТВЕННЫМ ПРЕДРАСПОЛОЖЕНИЕМ
Возникновение широко распространенных заболеваний, которые вносят наибольший вклад в заболеваемость, инвалидизацию и смертность населения,… Данные о роли наследственности в возникновении подобных заболеваний были… 1. Семейные исследования. Медицинским работникам хорошо известен факт накопления определенных заболеваний в пределах…ДИАГНОСТИКА НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Относится к числу основных в генетике человека и опирается на генеалогию -учения о родословных. Суть метода заключается в составлении родословной и… 1 этап.Составление родословной. Сбор сведений о семье начинается с пробанда - обычно это больной с изучаемым заболеванием. Дети одной родительской…Символы, используемые для составления родословной схемы
Близнецовый метод Это метод изучения генетических закономерностей на близнецах. Впервые он был… Монозиготные близнецы (МБ) образуются из одной зиготы, разделившейся на стадии дробления на две (или более) часта. С…Биохимические методы
Селективные программы предусматривают проверку биохимических аномалий обмена (моча, кровь) у пациентов, у которых подозреваются генные… основан на методах тонкослойной хроматографии мочи и крови для выявления… наличие у новорожденных судорог, комы, рвоты, гипотонии, желтухи.К просеивающим лабораторным методам относится определение сыворотки крови беременной веществ, получивших название сывороточных маркеров матери: альфа-фетопротеин (АФП), хорионический гонадотропин человека (ХГЧ), несвязанный эстриол (НЭ), ассоциированный с беременностью плазменный белок -А (РАРР-А).
АФП—это белок, вырабатываемый печенью плода во внутриутробном периоде его содержание меняется в течение беременности. АФП исследуется с целью выявления дефектов невральной трубки (анэнцефалия, зрта Ыйс1а). поликисгоза почек, омфалоцеле. врожденного нефроза, синдрома Дауна и др. Определяется в I триместре (10-14 недель) и во II триместре (16-20 недель). Повышение АФП выше 5-7 МОМ выявляется при пороках ЦНС. снижение уровня АФП характерно для синдрома Дауна.
В I триместре определяется также концентрация свободного ХГЧ и РАРР-А.
Во II триместре исследуется уровень концентрации в крови беременных женщин НЭ и общий и свободный ХГЧ.
Неинвазивные методы.
- срок и характер течения беременности. - толщину воротникового пространства в I триместре (ТВП). наличие или… - состояние хориона.Инвазивные методы
- структурные перестройки хромосом у супругов (у одного из супругов). - гетерозиготное носигельство. патологичского гена у обоих родителей при… наличие у родителей генного доминантного заболевания.Молекулярно-генетнческие методы
Это большая и разнообразная группа методов, предназначенная для выявления вариаций в структуре исследуемого участка ДНК (аллеля. гена, региона хромосомы) вплоть до расшифровки первичной последовательности оснований.
Основные этапы ДНК-диагностики.
а) выделение всей ДНК (тотальной или геномной) из клеток: б) накопление определенных фрагментов с помощью ПЦР. Источником геномной ДНК являются любые ядросодержащие клетки. Выделенная из клеток ДНК представляет собой весь геном…ОРГАНИЗАЦИЯ МЕДИКО-ГЕНЕТИЧЕСКОЙ СЛУЖБЫ
медико-генетическое консультирование (МГК): пренатальная диагностика: ДНК-диагностика (в настоящее время диагностируется более 40 наследственных… мониторинг врожденных пороков развития: преконцепционная профилактика: генотерапия (в стадии исследования). Уровни…Медико-генетическое консультирование
1. консультация врача-генетика как врачебное заключение: 2. специализированное учреждение здравоохранения Основная цель МГК - предупреждение рождения больного ребенка. Выделяют два типа консультирования:Периконцепционная профилактика врожденной и наследственной патологии
Основные положения периконцспционной профилактики: - беременность не должна быть случайной, она должна планироваться: - за 3-4 месяца до зачатия супруги должны убедиться в хорошем состоянии своего соматического, психического и…Симптоматическое лечение
Необходимо подчеркнуть, что симптоматическая терапия будет использоваться и в дальнейшем, наряду с самыми современными методами патогенетического и…Патогенетическое лечение
При патогенетических подходах к лечению наследственной патологии исходят из того, что у больных либо образуется аномальный белок (фермент), либо…Этиологическое лечение
Сложности этиологического лечения наследственных болезней очевидны, хотя уже имеются определенные возможности для их решения. Принципиально вопросы генной терапии у человека уже решены, т. с. на… В настоящее время лечение наследственных болезней представляет собой очень сложную задачу. К сожалению, далеко не…Хирургическое лечение
Трансплантация органов и тканей как метод лечения наследственных болезней в настоящее время находит широкое применение в медицинской практике. Пластическая хирургия широко применяется при лечении различных аномалий…– Конец работы –
Используемые теги: Генетика, наследственность, изменчивость0.061
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Генетика, Наследственность, Изменчивость
Что будем делать с полученным материалом:
Если этот материал оказался полезным для Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.158 сек.
Новости и инфо для студентов