рефераты конспекты курсовые дипломные лекции шпоры

- Раздел Медицина
- /
- Курс клинической генетики
Реферат Курсовая Конспект
Курс клинической генетики
Курс клинической генетики - раздел Медицина, Одесский Государственный Медицинский ...
ОДЕССКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ
УНИВЕРСИТЕТ
Кафедра клинической иммунологии, генетики и медицинской биологии
Курс клинической генетики
Бажора Ю.И., Шевеленкова А.В., Живац З.Н.,
Белоус О.Б., Чеснокова М.М., Николаевский В.В.
УЧЕБНОЕ ПОСОБИЕ К ПРАКТИЧЕСКИМ ЗАНЯТИЯМ
ПО КЛИНИЧЕСКОЙ ГЕНЕТИКЕ
Для иностранных студентов 5 курса
Медицинского факультета
ВведениеТема 1.
Цель занятия: Приобрести практические навыки обследования больного с целью… Студент должен знать:КОНТРОЛЬНЫЕ ВОПРОСЫ.
1. Задачи медико-генетического консультирования. Организация медико-генетической помощи населению.
2. Классификация наследственных болезней.
3. Синдромологический диагноз в клинической генетике.
4. Особенности клинических проявлений наследственной патологии.
5. Общие принципы клинической диагностики наследственных болезней.
6. Описание фенотипа больного с наследственной патологией.
7. Что такое микроаномалии развития, аномалии и пороки развития? Их значение в диагностике наследственной патологии.
8. Основные микроаномалии и пороки развития головы, лица, туловища, конечностей, кожи и ее производных.
9. Симптомы наследственной патологии в разные возрастные периоды.
10. Основные генетические термины: ген, генотип, фенотип, аллельные гены, гомозигота, гетерозигота, доминантный ген, доминантный признак, рецессивный ген, рецессивный признак, пробанд, сибс.
ПРАКТИЧЕСКАЯ РАБОТА
1. Проанализировать фенотип больного, назвать микроаномалии развития и пороки развития.
2. Проанализировать историю болезни , назвать признаки, указывающие на наследственную патологию.
3. На ксерокопиях и фотографиях изучить фенотипы больных с наследственными синдромами, назвать микроаномалии и пороки развития.
Основные теоретические сведения.
Задачи медико-генетического консультирования.
Установление точного диагноза наследственного заболевания. Определение типа наследования заболевания в данной семье; Расчет риска повторения болезни в семье;Классификация наследственных заболеваний.
Наследственные болезни - это болезни, обусловленные изменением генотипа (т.е. мутациями). Они не всегда передаются от родителей к детям. Многие наследственные заболевания не могут наследоваться (передаваться из поколения в поколение), так как они снижают жизнеспособность больного или вызывают бесплодие. Могут возникать у детей здоровых родителей в результате новой мутации. Например, у здоровых, как правило, родителей рождается ребенок с синдромом Дауна. С другой стороны, некоторые эндемичные заболевания наблюдаются у родителей и детей. Создается впечатление наследования, но заболевания не являются наследственными (например, эндемичный зоб).
Следует различать термины "наследственные" и "врожденные" заболевания. Врожденными называются любые болезни и пороки развития, проявляющиеся при рождении. Наследственные болезни не всегда врожденные. Первые симптомы могут проявляться в возрасте нескольких месяцев или даже лет (например, мукополисахаридоз, гепато - лентикулярная дегенерация Коновалова-Вильсона, хорея Гентингтона и др.) С другой стороны, некоторые врожденные пороки развития, вызванные воздействием тератогенного фактора, не являются наследственными.
"Семейные болезни" - заболевания, встречающиеся среди членов одной семьи. Семейными, как правило, являются рецессивные заболевания. Семейными могут быть и ненаследственные болезни, обусловленные одной и той же вредной профессией, семейными вредными привычками, неправильным питанием и др.
Генетическая классификация базируется на классификации мутаций и характере взаимодействия со средой. Всю наследственную патологию можно разделить на пять групп:
Моногенные болезни (генные болезни) - болезни, вызванные генными мутациями (ахондроплазия, фенилкетонурия, гемофилия и др.)
Хромосомные болезни - обусловленные изменением числа или структуры хромосом (синдром Дауна, кошачьего крика и др.)
Болезни с наследственной предрасположенностью (синоним: мультифакториальные)- болезни, развитие которых определятся взаимодействием измененного генотипа индивида и факторов окружающей среды (заячья губа, косолапость, гипертоническая болезнь, шизофрения и др.)
Генетические болезни соматических клеток - обусловлены соматическими мутациями (часто ретинобластома, опухоль Вильмса, некоторые спорадические случаи врожденных пороков и др.)
Болезни генетической несовместимости матери и плода (гемолитическая болезнь новорожденных, возникающая в результате несовместимости матери и плода по Rh-антигену и другим групповым антигенам).
Клиническая классификация основана на системном и органном принципе: нервно-мышечные, психические болезни, болезни опорно-двигательного аппарата, кожи, зубочелюстной области, крови и др.
Синдромологический диагноз в клинической генетике.
Большинство наследственных синдромом диагностируется только на основании характерной клинической картины. В большинстве случаев проводится… Известно большое количество наследственных заболеваний. Диагностику их… Необходимо не ограничиваться описанием фенотипа больного, а стремиться определить точный диагноз. Точный диагноз…Наследственной болезни.
Особенностью клинической генетики является то, что объектом исследования является не отдельный больной, а семья. Сбор сведений о семье начинается с… В настоящее время в медико–генетическую консультацию чаще обращаются семьи уже… Диагностика наследственных болезней, как правило, двухэтапная:Описание фенотипа больного с наследственной патологией.
Фенотип-это совокупность внешних и внутренних признаков организма. При описании фенотипа больного обращают внимание на пороки развития и… Врожденные пороки развития (ВПР) - стойкие морфологические изменения органа или всего организма, выходящие за пределы…Краткий перечень основных микроаномалий и пороков развития.
а) Изменение размеров больше чем на 10% от возрастной нормы (5 см) - микроцефалия (-5 см) или макроцефалия (+5 см). Гидроцефалия (водянка головного мозга) отличается фенотипически от… б) Форма черепа может быть обычная или аномальная – асимметричная. Наблюдается брахицефалия (относительное увеличение…Симптомы наследственной и врожденной патологии при объективном обследовании больного.
У новорожденных. 1. Врожденные пороки развития могут быть наследственными (моногенными,… 2. Недоношенность характерна для многих хромосомных синдромов.Основные генетические термины.
Генотип – система всех генов организма, содержащихся в диплоидном наборе хромосом. Фенотип –совокупность внешних и внутренних признаков организма. Аллельные гены –это гены, которые находятся в одинаковых локусах гомологичных хромосом и кодируют один и тот же…Тесты для проверки конечного уровня знаний.
1. Выберите один наиболее правильный ответ. Медицинская генетика изучает: А) клинические особенности наследственных болезней;ЛИТЕРАТУРА
Основная
1). Спадкові захворювання і прирождженні вади розвитку в перинаталогічній практиці /Запорожан В.М, Сердюк А.М, Бажора Ю.І та ін. др. – Київ, Здоров”я, 1997. – с.193 – 207.
Дополнительная
2). Педиатрия: пер. с англ./Гл. ред. Н.Н.Володин – М.: ГЭОТАР, 1996. – С. 205 – 240. 3). Медицина дитинства./За ред. П.С.Мощича. – Київ, Здоров”я, 1994. – С.238 –… 4). Л.В.Молодан, Е.Бугаева, О.О.Демина, И.В.Волчик. Описание фенотипа. – Харьков, 1998. – 49с.КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Что означают термины генеалогия, пробанд, сибсы?
2. Значение клинико-генеалогического метода.
3. Особенности сбора генеалогического анамнеза.
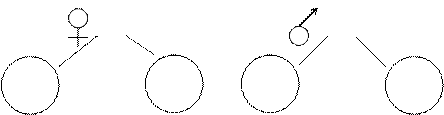
4. Генеалогическая символика. Правила графического изображения родословной.
5. Характерные особенности родословной при аутосомно-доминантном, аутосомно-рецессивном и сцепленном с полом типе наследования.
ПРАКТИЧЕСКАЯ РАБОТА
1. Собрать у больного генеалогический анамнез, составить и проанализировать родословную.
2. Составить свою собственную родословную.
3. Решить ситуационные задачи.
Основные теоретические сведения
Клинико-генеалогический метод основан на составлении и анализе родословной семьи. Это обязательный этап в обследовании больного с наследственной… Метод позволяет устанавливать следующее: 1) является ли признак единичным в семье или имеется несколько случаев данной патологии (семейный характер);Аутосомно-доминантный тип наследования.
Пример родословной с аутосомно-доминантным типом наследования:
 I
I
II
III
IV

1) Болезнь кодирует ген А, норму – а. Больные могут иметь генотип Аа или редко АА. У гомозигот заболевание проявляется в более тяжелой форме. При многих заболеваниях доминантный ген в гомозиготном состоянии обладает летальным действием.
2) Болеют мужчины и женщины одинаково часто.
3) Признак наследуется "по вертикали", т.е. встречается во всех поколениях.
4) У больного ребенка, как правило, болен один из родителей.
5) Если один из родителей болен, то вероятность рождения больного ребенка 50%.
 |
Р Аа х аа
гаметы А а а
F1 Aa aa
больны 50% здоровы 50%
Иногда при доминантном типе наследования родословные могут отличаться от классических. В 60-80% доминантный признак возникает из-за новой мутации у одного из родителей (чаще отцов). Риск новой доминантной мутации повышается с возрастом отца. Риск второй мутации, как правило, незначителен.
Иногда у здоровых родителей несколько детей могут иметь доминантный признак. В этом случае нужно думать о гонадном мозаицизме у одного из родителей. Мутация произошла в предшественниках половых клеток при образовании гонады. Многие половые клетки могут иметь мутантный ген. Риск рождения второго больного ребенка может быть достаточно высоким. Например, у двух здоровых родителей риск рождения второго ребенка с эктродактилией – 14%.
Вторая причина атипичных родословных – заболевание может проявиться не с момента рождения, а в более позднем возрасте (хорея Гентингтона чаще в 40-50 лет). Для некоторых доминантных генов характерна неполная пенетрантность и варьирующая экспрессивность.
Аутосомно-рецесивный тип наследования.
Пример родословной:
I
II
III
IV
Для этого типа наследования характерно следующее:
1) Болезнь кодируется рецессивным геном а, а норма – А. Больные имеют генотип аа, здоровые – АА или Аа. Для генов характерна полная пенетрантность. Варьирующая экспрессивность встречается редко.
2) Болеют мужчины и женщины одинаково часто.
3) Признак наследуется "по горизонтали", т.е. встречается у сибсов одного поколения.
4) Родители больного обычно здоровые гетерозиготы (Аа).
5) У двух гетерозиготных родителей риск рождения больного ребенка составляет 25%.
 Р Аа х Аа
Р Аа х Аа
гаметы А а А а
F1 AА Аa Аа aa
25%
6) Если оба родителя больны, то признак наследует 100% детей.
 Р аа х аа
Р аа х аа
гаметы а а
F1 aa
100%
7) Признаки часто встречаются при близкородственных браках, при многократном кровном родстве родословная может напоминать таковую при аутосомно-доминантном наследовании.
Атипичные родословные при известном рецессивном типе наследования могут быть объяснены мутацией у одного из родителей или унипарентной изодисомией.
1) Не исключена мутация у одного из родителей-гомозигот по доминантному гену:



 аа АА
аа АА
 |  | ||
ген А мутирует в рецессивный а
 а а
а а
аа
Возможна унипарентная изодисомия:
При нарушении второго деления мейоза в гамету может попасть две хроматиды (а††а). Зигота получит две хромосомы а††а из одной гаметы и †А – из другой. Хромосома †А теряется при первых делениях митоза. Происхождение двух хромосом от одного родителя доказывают результаты исследования хромосом с помощью методов дифференциальной окраски.
аа АА
 | |||
| |||
аа А
вначале образуется, по-видимому зигота Ааа, а затем ген
теряется, образуется зигота аа
При альбинизме и врожденной глухоте известны случаи рождения здоровых детей у двух больных родителей. Это объясняется генетической гетерогенностью заболеваний. У матери болезнь обусловлена одним мутантным геном (ааВВ), а у отца другим (ААbb).

 Р ааВВ х ААвв
Р ааВВ х ААвв
 гаметы аВ Ав
гаметы аВ Ав
F1 АaВв 100% здоровые
Сцепленное с полом наследование.
1) болеют примущественно мужчины; 2) никогда не наблюдается передача заболевания от мужчины к мужчине,… Р ХХ х ХУI
II
III
Доминантное сцепленое с Х-хромосомой наследование.
2) Если больная мать – гетерозиготная носительница патологического гена (ХАХа), то признак наследуют 50% дочерей и сыновей. Мать гетерозиготна по… Р ХАХа х ХаУ гаметы ХА Ха Ха УУ-сцепленый тип наследования.
Р ХХ х ХУg гаметы Х Х УgТесты для контроля конечного уровня знаний
1. Сибсы – это: а) родители пробанда; б) дети пробанда;Ответы на тесты конечного уровня знаний.
1. в, г 2.б, г, д 3.б 4. д 5. а, б, в, д 6.в. 7.г 8.б 9. а, б, в 10. а, б, в 11.а
Основная литература
1. Спадкові захворювання і природжені вади розвитку в перинатологічній практиці. /Запорожан В.М., Сердюк А.М., Бажора Ю.І. та ін. – Навч. посібник. – К.: Здоров’я, 1997. – с.122-126.
2. Резник Б.Я., Запорожан В.Н., Минков И.П. Врожденные пороки развития у детей. – Одесса, АОБАХВА, 1994. – с. 84-95.
3. Бердышев Г.Д., Криворучко И.Ф. Медицинская генетика. – К.: Вища школа, 1990. – с. 33-51.
Тема 4
Хромосомные болезни
Студент должен знать: 1. Характеристику кариотипа человека в норме. 2. Хромосомные и геномные мутациии.Практическая работа.
2. Проанализировать по фотографиям и ксерокопиям фенотипы больных с различными хромосомными болезнями. 3.Ознакомиться с методикой кариотипирования. Изучить на постоянных препаратах…Нормальный кариотип человека. Международная классификация хромосом человека.
Кариотип - диплоидный набор хромосом данного вида организма, характеризующийся постоянным числом, величиной и формой хромосом. В кариотипе человека 46 хромосом или 23 пары. Парные хромосомы называют гомологичными, они имеют одинаковою длину и форму, содержат аллельные гены. В состав хромосом входит 40 % ДНК, 40 % гистоновых белков и 20 % негистоновых белков. Комплекс всех химических веществ, входящих в состав хромосом, называется хроматином. Хромосомы могут находиться в клетках в двух структурных и функциональных состояниях – спирализованном и деспирализованном. В период интерфазы они находятся в деспирализованном состоянии. В спирализованном состоянии они находятся в период митоза. Максимальной спирализации хромосомы достигают в метафазе митоза. Метафазная хромосома состоит из двух хроматид, соединенных в области первичной перетяжки (центромеры). Некоторые хромосомы имеют вторичные перетяжки и спутники. Центромера делит хроматиду на два плеча. Короткое плечо принято обозначать буквой p а длинное буквой q .
Строение метафазной хромосомы.
 вторичная
вторичная 
 перетяжка p
перетяжка p 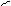

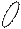





q
центромера
В 1960 г. английский генетик Патау предложил классифицировать хромосомы человека на основании относительной длины и положения центромеры (центромерного индекса). Центромерный индекс - отношение длины короткого плеча к длине всей хромосомы. В соответствии с центромерным индексом различают метацентрические (перетяжка посередине), субметацентрические (одно плечо длиннее второго), и акроцентрические хромосомы (с непропорционально очень коротким одним плечом).
 | |||||
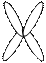 | |||||
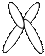 | |||||
метацентрические субметацентрические акроцентрические
В 1960 г на международном генетическом симпозиуме в Денвере (США) была принята Международная (Денверская) классификация хромосом человека. Основные принципы классификации разработал Патау. В классификации учтены длина и форма хромосом. Все пары аутосом нумеруют арабскими цифрами от 1 до 22 в порядке уменьшения их длины. Половые хромосомы обозначают латинскими буквами Х и У и располагают в конце раскладки. У женщин в норме половые хромосомы ХХ, а у мужчин ХУ.
Все пары аутосом распределяются на 7 групп в соответствии с длиной и формой хромосом. Группы обозначают латинскими буквами от А до G. Группы четко отличаются друг от друга.
Группа А (1,2,3 пары) самые длинные метацентрические (1,3) и субметацентрическая (2) хромосомы. Хромосома 1 - самая большая метацентрическая хромосома, центромера расположена посередине. Самой большой субметацентрической хромосомой является хромосома 2. Хромосома 3 почти на 20% короче хромосомы 1 и, следовательно, легко идентифицируется. Абсолютная длина от 11 мкм (1 пара) до 8,3 мкм (2 пара).
Группа В (4 и 5 пары) длинные субметацентрические хромосомы. Они не различаются между собой без дифференцированного окрашивания. Абсолютная длина 7,7 мкм.
Группа С (6 – 12 пары). Хромосомы среднего размера, субметацентрические. При стандартном (рутинном) окрашивании Х-хромосому нельзя отличить от других хромосом этой группы. Она по размерам сходна с хромосомами 6 и 7 пары. Абсолютная длина от 7,7 мкм (6 пара) до 5,8 мкм.
Группа D (13 - 15 пары). Эти акроцентрические хромосомы по форме сильно отличаются от всех других хромосом человека. Все три пары на коротком плече содержат спутники. Длина проксимальных участков коротких плеч варьирует, спутники могут отсутствовать, а могут быть очень большими, могут ярко флуоресцировать, а могут и не давать флуоресценции. Абсолютная длина от 4,2 мкм.
Группа Е (16 - 18 пары). Относительно короткие субметацентрические хромосомы. Абсолютная длина 3,6-3,5 мкм.
Группа F - (19 – 20 пары) маленькие метацентрические хромосомы. В препаратах при рутинной окраске они выглядят одинаково, но при дифференциальном окрашивании резко различаются. Абсолютная длина 2,9 мкм.
Группа G (21 – 22 пары) – две пары самых маленьких акроцентрических хромосом. На коротком плече имеют спутник. Изменчивость их коротких плеч так же значительна, как и в хромосомах группы D. Абсолютная длина 2,9 мкм.
Y-хромосома маленькая акроцентрическая хромосома длиной 2,8 мкм. Обычно (но не всегда) больше, чем хромосомы группы G, и хроматиды ее длинного плеча, как правило, лежат параллельно одна другой. Этим она отличается от хромосом группы G, у которых хроматиды длинных плеч образуют широкий угол.
Х хромосома сходна при рутинном окрашивании с хромосомами гриппы С, отличается при использовании дифференцированного окрашивания. Х - хромосома субметацентрическая, длиной 6,8 мкм.
Классификация хромосомных болезней.
В настоящее время описано более 1000 болезней. Около ста из них имеют четкую клиническую картину и называются синдромами. Все хромосомные болезни можно разделить на три группы в зависимости от характера изменения кариотипа.
Хромосомные болезни.
 |  | ||||
 | |||||
Полиплоидии Заболевания, связанные Заболевания, связанные
с изменением числа и с изменением числа и
структуры аутосом структуры половых
хромосом.
В зависимости от процента пораженных клеток различают полные хромосомные болезни и мозаичные. Полные являются следствие генеративной мутации у родителей (т.е. мутации происходят при образовании половых клеток у родителей). Все клетки эмбриона имеют измененный кариотип. Мозаичные являются следствием соматической мутации, которая возникает у самого эмбриона в процессе эмбрионального периода развития. Поэтому часть клеток больного имеют нормальный набор хромосом, а часть клеток измененные.
Выделяют также спорадические хромосомные болезни (следствие новой мутации) и наследуемые (наследуются от родителей со сбалансированной мутацией или от родителей с хромосомной болезнью). Описаны случаи рождения детей у больных с синдромами Клайнфельтера, полисомии Х у женщин, полисомии У у мужчин, у женщин с синдромом Дауна . Мужчины с синдромом Дауна бесплодны, т.к. у них нарушается сперматогенез.
Этиология хромосомных заболеваний.
Хромосомные мутации -это изменения в структуре хромосом. Являются, как правило, следствием неравного кроссинговера при мейозе. Выделяют следующие… 1. Делеция или нехватка. Утрачен участок хромосомы. A B C Д Е F G H Делеция G H А В С Д Е FГеномные мутации.
1) полиплоидию, 2) гетероплоидию (анеуплоидию). Полиплоидия– увеличение числа хромосом на величину кратную гаплоидному набору (3n, 4n...). У человека описана…Норма (2n) ХХ ХХ ХХ
Мы видим по две хромосомы каждой пары в гипотетической клетке с 6 хромосомами или 3 парами. а) Моносомия - отсутствие одной хромосомы (2n-1) ХХ ХХ Х-Причины формирования геномных мутаций.
Схема нормального мейоза
У мужчин у женщин
23,Х 23,У 23,Х 23,ХНерасхождение 21 хромосомы при мейозе у женщины.
гаметы 21,21 0 21Запись нормального и измененного кариотипа человека.
Нормальный кариотип человека: 46,ХХ - женщина; 46, ХУ - мужчина. Кариотип при полиплоидии:Патогенез хромосомных болезней.
От 35 до 50% (сейчас пишут до 70%) зародышей человека погибают на стадии бластоцисты, т.е. до имплантации. У большого процента изних наблюдаются… Если аборт происходит в первые 2-4 недели, то хромосомный дисбаланс… Если нарушения эмбрионального развития совместимы с жизнью, то рождается ребенок с пороками развития.КЛИНИКО-ЦИТОГЕНЕТИЧЕСКАЯ ХАРАКТЕРИСТИКА НАИБОЛЕЕ РАСПРОСТРАНЕННЫХ ХРОМОСОМНЫХ БОЛЕЗНЕЙ
Общие признаки: В ПЕРИОД БЕРЕМЕННОСТИ. 1. Угрозы прерывания беременности.Синдром Патау (трисомия 13).
Минимальные диагностические признаки: микроцефалия, полидактилия, расщелина губы и неба, трисомия 13 хромосомы.
Синдром Патау встречается в основном в двух цитогенетических вариантах: в виде простой трисомии и робертсоновских транслокаций Д/13, фенотипически… Основные диагностические признаки при синдроме Патау: черепно-лицевые… Прогноз: около 2/3 больных погибает в перинатальном периоде от дыхательных расстройств, приступов апноэ, судорог.…Синдром трипло-Х, трисомия Х, тетрасомия Х, пентасомия X/.
Тетрасомия Х /48,ХХХХ / и пентасомия Х /49,ХХХХХ/ встречаются крайне редко. Клиническая картина трисомии Х вариабельна – от практически здоровых… Женщины с кариотипом 47, ХХХ имеют в основном нормальное физическое и психическое развитие. Чаще всего таких индивидов…Диагностика хромосомных заболеваний
1. Метод кариотипирования. 2. Метод определения полового хроматина. Метод кариотипирования.Показания для кариотипирования.
1. Дети с фенотипом, характерным для определенного хромосомного заболевания. 2. Дети с множественными врожденными пороками развития или с признаками… 3. Дети с отставанием в умственном развитии неясной этиологии.Медико-генетическое консультирование.
1. У родителей нормальный кариотип. В этом случае риск для сибсов пробанда оценивается по эмпирическим данным для каждого типа аномалии с учетом… Суммарный популяционный риск рождения детей с трисомиями (синдромы Дауна,…Тесты для самоконтроля.
1. Выберите два правильных ответа. Какие мутации относятся к геномным: А) инверсии, транслокации, дупликации, делеции;Литература.
Основная.
1. Спадкові захворювання і природжені вади розвитку в перинатологічній практиці. (Запорожан В.М., Свердюк А.М., Бажора Ю.І. та інші) – навчальний посібник.- К.: Здоров,я – 1997. – С. 78-94, 132-137, 228-241.
2. Резник Б.Я., Запорожан В.Н., Минков И.П. Врожденные пороки развития у детей. Одесса. – 1994. – С.323-358.
Дополнительная.
1. Захаров А.Ф., Бенюш В.А., Кулешов Н.П. и др. Хромосомы человека. (Атлас). – М.: Медицина, 1982. – С.8-13, 44-51.
2. Бочков Н.П. Клиническая генетика. – М.: Медицина. – 1997. – 288с.
3. Бердышев Т.Д., Криворучко И.Ф. Медицинская генетика. – К.: Вища школа. – 1990. – 336с.
4. Наследственные синдромы и медико – генетическое консультирование (Козлова С.И., Демикова Н.С., Семакова Е., Блинникова О.Е.) М.: Медицина, 1996. – 416с.
5. Тератология человека. Руководство для врачей. (Под редакцией Г.И. Лазюка). –М.:Медицина, 1991. – 480с.
Тема 2
Моногенные болезни и синдромы
Студент должен знать: 1. Классификацию моногенных болезней. 2. Общую характеристику моногенных болезней с разными типами наследования.КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Этиология и патогенез моногенных болезней. Классификация генных мутаций.
2. Классификация моногенных болезней и синдромов.
3. Моногенные болезни и синдромы с аутосомно-доминантным типом наследования: ахондроплазия, синдром Марфана, акроцефалосиндактилия (синдрома Апера). Клиника, диагностика, ведение и лечение больных, пренатальная диагностика, медико-генетическое консультирование, определение генетического риска.
4. Моногенные болезни и синдромы с аутосомно-рецессивным типом наследования: альбинизм, фенилкетонурия, муковисцидоз, гипотиреоз.
5. Моногеныые болезни и синдромы со сцепленным с Х-хромосомой типом наследования: мышечная дистрофия Дюшенна, фосфат-диабет, гемофилия, ангидротическая эктодермальная дисплазия.
6. Наследственные нарушения обмена веществ, их классификация. Симптомы, указывающие на наследственное нарушение обмена веществ.
7. Болезни накопления. Гликогенозы. Мукополисахаридозы.
8. Биохимические методы диагностики моногенных болезней. Скрининг новорожденных.
9. Методы ДНК-диагностики.
10. Пренатальная диагностика моногенных болезней и синдромов.
ПРАКТИЧЕСКАЯ РАБОТА:
1. Обследовать больного с моногенной наследственной болезнью, указать характерные симптомы, составить родословную и установить тип наследования.
2. Составить план обследования больного с моногенной болезнью.
3. Проанализировать данные лабораторного обследования больного фенилкетонурией и муковисцидозом.
4. Проанализировать на фотографиях и ксерокопиях фенотипы больных с моногенными болезнями.
Основные теоретические сведения.
Ген в узком смысле слова – это участок ДНК, в котором закодирована информация о строении одного белка. Выделяют следующие виды генных мутаций: 1. Замена нуклеотидов на комплементарный или некомплементарный. Например, при…Генетическая классификация моногенных заболеваний
С учетом типа наследования
Аутосомно- Аутосомно- Сцепленные Митохондриальные доминантные рецессивные с полом (АД) (АР)Патогенетическая классификация
 |  |  | |||
Наследственные Врожденные Комбинированные
болезни обмена пороки разви- состояния
веществ (ферменто- тия
патии)
В настоящее время разработаны более сложные патогенетические и клинические классификации. Согласно этой классификации выделяют следующие группы моногенных болезней.
1. Врожденные пороки развития (микроцефалия, эктродактилия).
2. Ферментопатии – нарушения выработки ферментов, которые проявляются нарушениями обмена веществ или эндокринопатиями (фенилкетонурия).
3. Болезни накопления – разновидность ферментопатий (мукополисахаридоз).
4. Болезни, связанные с нарушением мембранного транспорта различных веществ в клетку (разновидность ферментопатий). Например, муковисцидоз.
4. Патология синтеза структурных белков (чаще с аутосомно-доминантным типом наследования)- болезни соединительных тканей (синдром Марфана, несовершенный остеогенез и др.)
5. Нарушения структуры и функций белков крови- гемоглобина, факторов свертывания крови, транспортных белков (серповидно-клеточная анемия, гемофилия).
6. Наследственные болезни системы иммунитета (дефицит аденозиндезаминазы А).
7. Болезни, связанные с нарушением репарации ДНК и болезни хромосомной нестабильности (пигментная ксеродерма, синдром Блума).
8. Митохондриальные и пероксисомные болезни (атрофия зрительного нерва Лебера).
9. Болезни, обусловленные унипарентной дисомией (наследованием двух хромосом от одного родителя) - синдромы Рассела-Сильвера, Беквита-Видеманна.
10. Болезни с неизвестной этиологией (синдром Корнелии-де-Ланге и др.)
Для медико-генетического консультирования очень важно знать тип наследования.
Это позволяет рассчитать риск рождения больного ребенка. Поэтому в дальнейшем при изложении материала мы будем придерживаться генетической классификации.
Общая характеристика моногенных болезней
2). Генетическая гетерогенность или генетический полиморфизм. Некоторые болезни могут быть обусловлены в разных семьях мутациями совершенно разных… 3). Клинический полиморфизм может быть также обусловлен генетическим… 4).Прогредиентное течение характерно для болезней накопления и других ферментопатий. Прогредиентность присуща не всем…Аутосомно – доминантные заболевания
Р Аа х аа гаметы А,а а F1 Аа, ааАхондроплазия
Синдром Марфана
Заболевание обусловлено мутацией гена, кодирующего белок соединительной ткани фибриллин. Ген локализован в длинном плече 15 хромосомы (15q). При… Все это приводит к гиперрастяжимости соединительной ткани. Наблюдается… Опорно-двигательная система: длинные и тонкие пальцы (арахнодактилия) и конечностей, высокий рост, сколиоз и…Моногенные болезни и синдромы с аутосомно-доминантным типом наследования
Аутосомно-рецесивные заболевания
Мужчины и женщины болеют одинаково часто. Родители больного, как правило здоровы, но являются гетерозиготными носителями патологического гена (Аа).… Р Аа х АА Гаметы А,а А,аПримеры врожденных пороков развития с аутосомно-рецессивным типом наследования
| Название синдрома | Минимальные диагностические признаки |
| Анофтальмия Голопроз энцефалия семейная алобарная Микроцефалия (первичная) | Отсутствие глаз Неразделение переднего мозга на полушария, срединная расщелина губы и неба, гипотелоризм Уменьшение объёма головы, уменьшение массы и размеров мозга, умственная отсталость. Могут быть вторичные микроцефалии при многих генных, хромосомных и тератогенных синдромах. |
Наследственные нарушения обмена веществ – самая многочисленная группа аутосомно-рецессивных заболеваний. Чаще они являются следствием мутации генов, кодирующих ферменты (ферментопатии или энзимопатии).
Врожденные нарушения обмена веществ в основном наследуются по аутосомно-рецессивному типу, реже сцепленно с Х – хромосомой.
Классификация и примеры ферментопатий
1. Нарушение обмена аминокислот (аминоацидурия) -фенилкетонурия, альбинизм.
2. Нарушение обмена углеводов - галактоземия, гликогенозы.
3. Нарушение обмена липидов - гиперхолестеринемия, болезнь Тея-Сакса.
4. Нарушение обмена пуринов - синдром Леша-Нихана.
5. Нарушение обмена аммониевых соединений - дефицит орнитинтранскарбамилазы.
6. Нарушения обмена металлов - болезнь Вильсона – Коновалова (гепатолентикулярная дегенерация).
7. Нарушение обмена органических кислот (органических ацидемий) - пропионовая, метилмалоновая, изовалериановая ацидемия.
8. Нарушение синтеза гормонов - гипотиреоз, врожденная гиперплазия надпочечников.
9. Нарушение транспорта хлоридов - муковисцидоз.
10. Нарушение билирубинового обмена - синдромы Дубина-Джонсона, Криглера-Найяра.
Примеры наследственных нарушений обмена веществ.
| Название заболевания | Частота | Локализация гена | Минимальные диагностические признаки | ||||
| Нарушение обмена аминокислот | |||||||
| Фенилкетонурия – нарушение обмена аминокислоты фенилаланина | 1:8000 1:6000 | 12q | Нарушение процесса гидроксилирования фенилаланина в тирозин, накопление фенилаланина и его метаболитов в крови и выделение их с мочой. Повышенная возбудимость, гиперрефлексия, повышенный тонус мышц, “мышиный запах”. Позже умственная отсталость, микроцефалия, уменьшение пигментации кожи, волос, радужки. При раннем назначении диеты нормальное развитие. | ||||
| Альбинизм – нарушение обмена аминокислоты тирозина | 1:39000 | 11q | Тотальная депигментация кожи, волос, глаз; светобоязнь, красный зрачковый рефлекс. Радужка обычно серо-голубая, но может быть розовая. | ||||
| Нарушение обмена углеводов | |||||||
| А) Дефекты ферментов, расщепляющих моносахариды и дисахариды. | |||||||
| Галактоземия –нарушение обмена моносахарида галактозы (входит в молочный сахар) | 1:35000 1:150000 | 9p | Появление первых симптомов после приёма молока. Отставание психомоторного развития, катаракта, гепатомегалия, желтуха. Галактозурия. При раннем назначении диеты (специальные смеси без лактозы) нормальное развитие. | ||||
| Б) Дефекты ферментов, расщепляющих полисахариды. | |||||||
| Мукополисахаридозы– нарушение катаболизма гликозаминогликанов (ГАГ), болезнь накопления. Описано 14 типов (АР и один ХР). | СиндромГурлер 4р Xq3.5.7.22 и др. | Поражение ЦНС и соединительной ткани. Поражение скелета (тугоподвижность и деформации суставов, деформации позвоночника, отставание в росте), отложение ГАГ в печени, селезёнке, миокарде. Поражение ЦНС (снижение интеллекта), глаз (катаракта), слуха (тугоухость), сердечно-сосудистой системы (кардиомегалия, поражение клапанов). Грубые черты лица. | |||||
| Гликогенозы – нарушение катаболизма гликогена, болезнь накопления. Описано 11 типов. Тип 1 –болезнь Гирке. | Отложение гликогена в различных тканях. Отставание в росте, гепатомегалия, гипогликемия, мышечная гипотония, кардиомегалия, “кукольное лицо”. | ||||||
| Нарушение обмена липидов | |||||||
| Семейная гиперхолестеринемия – нарушение синтеза рецептора к ЛПНП. АД тип наследования. GМ2 – ганглиозидоз (болезнь Тея-Сакса) -нарушение катаболизма ганглиозидов. Болезнь накопления | Аа 1:200 1:500 аа 1: 106 1:500 среди евреев | 15q | Ранний атеросклероз, инфаркты, инсульты, ксантомы на коже, в сухожилиях. У гомозигот (АА) более тяжелая форма чем у гетерозигот (Аа). С 4-5 мес. отставание в психомоторном развитии, симптом “вишневой косточки” на глазном дне. Слепота, глухота, идиотия. Смерть в 3-4 года. Дефицит гексозаминидазы А в сыворотке крови и тканях | ||||
| Нарушение обмена пуринов | |||||||
| Синдром Леша-Нихана – тип наследования ХР. | Xq | Умственная отсталость, хореоатетоз, аутоагрессия с самоповреждениями (кусают пальцы, язык и т.д.). Повышение уровня мочевой кислоты в крови и моче. | |||||
| Нарушение обмена меди | |||||||
| Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) | 13q | Снижение концентрации церулоплазмина в плазме, отложение меди в печени, затем в головном мозге, почках. Гепатит (напоминающий хронический активный гепатит), гепато- и спленомегалия, поражение ЦНС (дисфагия, дизартрия, гиперкинезы). Кольцо Кайзера-Флейшера на радужке (отложение меди). Лечение даёт хороший эффект. | |||||
| Нарушение транспорта хлоридов | |||||||
| Муковисцидоз (кистофиброз) – нарушение транспорта хлоридов в клетки, выделение густой слизи | 1:1600 | 7q | Рецидивирующие пневмонии, нарушение выделения ферментов поджелудочной железы, нарушение всасывания в кишечнике. У мужчин бесплодие. Повышение концентрации ионов натрия и хлора в поте. | ||||
| Нарушение синтеза гормонов | |||||||
| Гипотиреоз – разнородная по причинам группа заболеваний. | 1:3500 1:4000 | Резкое отставание в психомоторном развитии, характерный внешний вид (короткая шея, широкий нос, Запавшая переносица, узкие глазные щели, отеки век, макроглоссия, редкие волосы). Затянувшаяся желтуха, грубый голос, брадикардия, гипотермия. Снижение уровня тироксина, повышение ТТГ. При своевременном лечении хороший эффект. | |||||
| Адреногенитальный синдром (врожденная гиперплазия надпочечников) – нарушение биосинтеза стероидных гормонов. Описано 5 типов, в 90% - дефицит 21 – гидроксилазы. | 1:5000 | 6p | Сольтеряющая форма обусловлена дефицитом минералокортикоидов. Симптомы: рвота, тахикардия, сонливость, дегидратация, гипонатриемия, гиперкалиемия, ацидоз. Вирильная форма у девочек проявляется маскулинизацией половых органов (гипертрофия клитора, срастание губно-мошоночных складок), у мальчиков с 5-7 лет преждевременное половое созревание. Заместительная терапия гормонами дает хороший эффект. | ||||
Общие симптомы наследственных нарушений обмена веществ.
1. В семейном анамнезе смерть детей в раннем возрасте. 2. Нормальное течение настоящей беременности. Дети, как правило, рождаются… Б). Клинические признаки.Задержка психомоторного развития у детей раннего возраста, умственная отсталость у старших характерны для аминоацидурий (фенилкетонурии), болезней накопления (мукополисахаридозов), для гипотиреоза, нарушения обмена углеводов (галактоземии) и др.
6. Судороги неясного генеза и другая неврологическая симптоматика (вялость, раздражительность, гипо – и гипертонус мышц, вялое сосание и др.) наблюдаются при аминоацидуриях, нарушении обмена органических кислот.
7. Эпизоды обмороков, переходящих в кому.
8. Катаракта – один из симптомов галактоземии, мукополисахаридоза
9. Отсутствие эффекта от обычной терапии, прогрессирование симптомов без убедительных доказательств инфекции или поражения ЦНС.
В). Лабораторные показатели.
1. Ацидоз с анионным провалом развивается при аминоацидуриях.
2. Гипераммониемия обычно связана с нарушениями обмена мочевины и органических кислот.
3. Стойкие отклонения от нормы других показателей – гипогликемия, кетонурия, положительная реакция мочи на редуцирующие вещества, гипербилирубинемия.
Г) Другие симптомы – непереносимость некоторых продуктов, кожные проявления (опрелости, дерматиты, нарушения пигментации), деформация скелета.
Указанные симптомы появляются, как правило, после периода благополучия от одного до нескольких дней. Беременность, как правило, протекает благополучно, дети рождаются доношенными. Всё это объясняется тем, что в период беременности нарушения метаболизма у плода компенсируется за счет организма матери.
Очень часто у детей раннего возраста выставляют диагнозы сепсиса, перинатальной энцефалопатияи, сердечно-легочной патологии, пилоростеноза, асфиксии, печеночной недостаточности. В пользу врожденного нарушения обмена веществ будут свидетельствовать отсутствие убедительных доказательств инфекции или поражения ЦНС, отсутствие эффекта от обычной терапии, стойкие отклонения от нормы лабораторных показателей.
Фенилкетонурия
В основе классической фенилкетонурии лежит блок фермента фенилаланингидроксилазы, вследствии чего фенилаланин крови не превращается в тирозин.… Дети с ФКУ рождаются здоровыми, первые симптомы появляются с двухмесячного… Диагноз ставится на основании клинической картины и результатов биохимического исследования мочи (фенилпировиноградная…Что такое болезни накопления?
К болезням накопления относятся мукополисахаридозы, сфинголипидозы, гликогенозы. Они часто обусловлены мутациями генов лизосом, поэтому называются… Мукополисахаридозы. Для мукополисахаридоза характерно отложение в различных тканях организма полимерных углеводов, называемых…Сцепленное с Х-хромосомой наследование.
Сцепленные с Х-хромосомой гены могут быть доминантными и рецессивными. Рецессивные сцепленные с Х-хромосомой гены обозначают Хh. Эти заболевания… Женщины чаще всего являются здоровыми гетерозиготными носительницами (ХH Хh) и… При наличии клинической картины рецессивного заболевания сцепленного с Х-хромосомой у девочек следует также исключить…Диагностика моногенных болезней.
1). Портретная диагностика – для диагностики моногенных синдромов, проявляющихся пороками развития. 2). Биохимические методы – для диагностики наследственных нарушений обмена… 3). ДНК- диагностика для ферментопатий и др.Первичный (скрининг) и 2) уточнение диагноза.
Основная цель скрининга заключается в отборе индивидов для последующего уточнения диагноза. Скрининг может быть селективным и массовым.
Селективный биохимический скрининг проводится среди групп детей и взрослых с подозрением на наследственный дефект обмена веществ. Иными словами, речь идет о “просеивании” специальных контингентов детей или взрослых, среди которых можно ожидать значительное увеличение частоты больных с наследственными болезнями обмена.
Показания для селективного скрининга:
1) задержка психомоторного равития у детей раннего возраста , умственная отсталость у детей старшего возраста;
2) неврологические нарушения (судороги, снижение мышечного тонуса, спастические парезы);
3) диспепсические явления, непереносимость отдельных продуктов и лекарств, нарушение вскармливания;
4) нарушение физического равития детей (задержка в прибавке массы тела, неправильный рост, деформация костей туловища и конечностей);
5) другие симптомы ферментопатий (катаракта, нарушение слуха, зрения, специфический цвет и запах мочи, кожные проявления и др. ).
В селективных программах могут использоваться качественные и полуколичественные методы. В целом одна порция мочи или крови исследуется с помощью приблизительно 30 различных реакций. Они позволяют обнаружить большие группы отклонений (например, тонкослойная хроматография –ТСХ- мочи и крови позволяет диагностировать наследственные нарушения обмена аминокислот, олигосахаридов, мукополисахаридов). Для уточнения диагноза часто необходим второй этап, на котором используются более точные методы диагностики.
Массовый скрининг – это массовое обследование всех новорожденных с целью раннего выявления на доклинической стадии заболевания, которое можно лечить. Раннее лечение предупреждает развитие клинических признаков болезни.
Основные требования к скрининговым программам для массового скрининга.
1. Частота в популяции 1:10000 (иногда скринируются менее распространенные заболевания).
2. Заболевание без своевременного лечения приводит к тяжелым нарушениям здоровья, ранней инвалидизации.
3. Должны существовать способы профилактического лечения.
4. Методы диагностики должны быть высоко чувствительными, не должны давать ложно отрицательных результатов.
5. Затраты на скрининг должны быть меньше затрат на лечение и содержание больных – инвалидов.
Программа обязательно включает в себя следующие этапы: I) взятие биологического материала для исследования у всех новорожденных и доставка материала в диагностическую лабораторию; 2) лабораторная просеивающая диагностика; 3) уточняющая диагностика всех случаев с положительными результатами при просеивании; 4) лечение больных и их диспансеризация с контролем за ходом лечения; 5) медико-генетическое консультирование семьи.
Таким образом, программы массового скрининга на наследственные болезни, поддающиеся профилактическому лечению, могут учреждаться только в рамках государственного или регионального здравоохранения. Разработаны программы массового скрининга фенилкетонурии, галактоземии, гипотиреоза, адрено - генитального синдрома, муковисцидоза.
Скрининг на фенилкетонурию.
На 3-5 сутки новорожденных берут из пятки кровь на специальную фильтровальную бумагу (карточки Гатри). Кровь высушивают, образец пересылают в лабораторию, занимающуюся скрининговыми исследованиями.
Для скрининга могут использовать 3 методики: 1) тест Гатри, 2) тонкослойная хроматография аминокислот крови, 3) флюориметрический метод. Тест Гатри микробиологический. В планшеты высевают бактерии Bacillus subtilis на среду с антиметаболитом фениланина. На этой среде бактерии не растут. Из пятна высушенной крови вырезают кружки (диски) диаметром 2 мм. Кружки раскладывают на питательную среду. В крови есть фенилаланин, поэтому вокруг пятна с кровью появляется рост бактерий. Интенсивность роста зависит от количества фенилаланина. Метод Гатри качественно-полуколичественный. Позволяет определить концентрацию ФА выше 4 мг %.
Метод тонкослойной хроматографии (ТСХ) также качественно-полуколичественный, определяет концентрацию ФА выше 4 мг%, технически относительно прост и дешев, позволяет диагностировать не только ФКУ, но и другие аминоацидурии. В Одесском ММГЦ метод ТСХ аминокислот крови используют не только для диагностики ФКУ и других аминоацидурий, но и для контроля эффективности лечения.
Флюориметрический метод – бумагу с кровью помещают в специальный раствор и добавляют краситель нингидрин. Нингидрин с фенилаланином дает сиреневую окраску. Интенсивность окраски зависит от количества фенилаланина и определяется с помощью специального прибора – флюороскана. Если не осуществляется массовый скрининг в роддоме, то можно использовать тест с треххлорным железом (FeCl3). Его проводят на 2-3 месяце жизни. К моче добавляют HCl и FeCl3. При наличии фенилаланина моча имеет зеленную окраску. Этот метод является количественым, наиболее точным, позволяет определить концентрацию ФА.
Скрининг гипотиреоза и адреногенитального синдрома (АГС) основан на определении уровня гормонов в крови.
Для скрининга галактоземии используется микробиологический или биохимический тесты, определяющие содержание галактозы в крови.
Скрининг муковисцидоза основан на определении активности трипсина в крови или содержание альбумина в меконии.
Необходимо помнить о том, что процедура скрининга не обеспечивает окончательного диагноза, так как для скрининга используются высокочувствительные реакции, которые в ряде случаев могут давать ложноположительные реакции. Необходим второй этап - повторное обследование и подтверждение диагноза.
Пренатальная диагностика моногенных болезней и синдромов.
1). УЗИ – для диагностики пороков развития. Оптимальные сроки для скрининговых исследований 1-й раз в 16-18 недель, 2-й раз в 24-26 недель. По… Врожденные пороки, которые диагностируются с помощью УЗИ (моногенные, мультифакториальные, теротогенные) Система или орган Пороки. ЦНС …Тесты для самоконтроля.
1. Укажите вероятность повторного рождения больного ребенка у супругов, иемющих… А) 50%;Задания для самостоятельной работы студентов на практическом занятии.
1. Обследовать больного с моногенным нследственным заболеванием, указать хараткерные симптомы, составить родословную и установить тип наследования.
2. Составить план обследования больного с моногенным заболевнаием.
3. Проанализировать на фотографиях и ксерокопиях фенотипы больных с моногенными заболеваниями и синдромами.
4. Проанализировать данные лабораторного обследования больного на фенилкетонурию и муковисцидоз.
Ответы на тесты и задания для проверки конечного уровня знаний.
1. г 2. б 3. б 4. а, в, г 5. а, в 6.в 7 .б 8.в 9.б 10.в 11. б 12. б 13. г 14. г 15. б 16. г 17. г 18. г 19. б 20. А-2,Б-5,В-4,Г-1,Д-3,Е-6,Ж-8,З-7.
Литература.
Основная:
1. Спадкові захворювання і природжені вади розвитку в перинатологічній практиці. (Запорожан В.М., Сердюк А.М., Бажора Ю.І. та інші) – навчальний посібник.- К.: Здоров’я – 1997. – С.75-78, 241-274, 141-148.
2. Резник Б.Я., Запорожан В.Н., Минков И.П. Врожденные пороки развития у детей. Одесса. – 1994. – С. 153-166, 372-372, 377-378.
3. Текст лекции.
Дополнительная:
1. Бочков Н.П. Клиническая генетика. – М.: Медицина. – 1997. – 288с.
2. Бердышев Т.Д., Криворучко И.Ф. Медицинская генетика. – К.: Вища школа. – 1990. – 336с., ил.
3. Наследственные синдромы и медико – генетическое консультирование (Козлова С.И., Демикова Н.С., Семанова Е., Блинникова О.Е.) М.: Медицина, 1996. – 322, 416 ил..
4. Тератология человека. Руководство для врачей. (Под редакцией Г.И. Лазюка). –М.:Медицина, 1991. – 480с.
Тема 5
Методы ДНК – диагностики.
Студент должен знать: 1. Строение генома человека. 2. Основные термины генетики: ген, генотип, геном.ПРАКТИЧЕСКАЯ РАБОТА.
1. Ознакомиться с работой молекулярно-генетической лаборатории.
2. Ознакомиться с этапами метода ДНК- диагностики с использованием полимеразной цепной реакции.
3. Ознакомиться с оценкой результатов молекулярно-генетического исследования.
Основные теоретические сведения.
С 1900 г. – года официального рождения – генетика прошла путь от открытия количественных закономерностей наследственности законов Менделя и… Наследственная (генетическая) информация – это информация обо всей программе… Впервые ДНК открыл в 1868г. Мишер в ядрах лейкоцитов, благодаря чему эти вещества и получили название нуклеиновых…Строение и функции ДНК.
В ДНК две полинуклеотидные цепи, свернутые в форме спирали. Толщина ДНК 2нм, в одном витке спирали 10 пар нуклеотидов, длина одного витка 3,4нм. Азотистые основания располагаются в двух цепях по принципу комплементарности.… ДНК находится в ядре клетки (входит в состав хромосом) и в митохондриях (у растений также и в хлоропластах). Главная…Строение и функции РНК.
Отличия РНК от ДНК: 1) состоит из одной полинуклеотидной цепи; 2) в состав нуклеотида входит рибоза;Ген, генетический код и его свойства.
Порядок чередования нуклеотидов в ДНК кодирует определенную последовательность аминокислот в полипептиде (первичной структуре белка). Способ… В 1954 г. А. Гамов предположил, а в 1961 г. Ф. Крик доказал, что генетический… Строение всех триплетов было открыто благодаря работам М. Ниренберга, Дж. Маттеи (1961) и С. Очоа (1962).Классификация генов.
Указанное выше определение гена является узким. В широком смысле слова ген – это участок ДНК, содержащий определенный объем генетической информации и кодирующий РНК и/или полипептид. Определенные участки ДНК не кодируют какие – либо структуры, а регулируют работу структурных генов. Эти участи называются регуляторными генами. Следовательно, гены можно подразделить на две большие группы:
1) структурные гены (кодируют белки, рРНК, тРНК);
2) регуляторные (промоторы, операторы, терминаторы, энхансеры и др.)

 Классификация генов
Классификация генов
Структурные регуляторные
Белки рРНК тРНК промоторы операторы терминаторы
Что такое геном и генотип? Характеристика генома человека.
Система всех генов организма в гаплоидном наборе хромосом называется геномом. В настоящее время сюда прибавляют гены ДНК митохондрий. Система всех генов данного организма называется генотипом (у человека в… Количество ДНК в геноме обозначают буквой С и измеряют в парах нуклеотидов (п.н.). У человека в геноме 3 х 109 п.н.…Программа «Геном человека».
Программа включает три направления: 1) картирование генов человека и выяснения нуклеотидной последовательности… 2) структурно - функциональное изучение генома;Каталог генов и генных болезней В.Мак-Кьюсика.
«Менделевское наследование у человека: каталог человеческих генов и генетических болезней» – так называется энциклопедия, которую издает Мак-Кьюсик… 1. –АД (аутосомно-доминантный) 2. –АР (аутосомно-рецессивный)Молекулярно-генетические методы. Использование в медицине.
Молекулярно-генетические методы (методы ДНК –диагностики) – это большая и разнообразная группа методов, предназначенная для выявления вариаций в… 1. Для диагностики моногенных и мультифакториальных наследственных… 2. Для пренатальной диагностики моногенных болезней или пола плода (в случаях Х – сцепленного наследования).Этапы ДНК-диагностики с использованием полимеразной цепной реакции
2. Выделение ДНК.Для исследования методом ПЦР пригодны даже небольшие фрагменты слабоочищенной ДНК. Как правило, ДНК из биологического материала при… Полимеразная цепная реакция (ПЦР) позволяет избирательно амплифицировать… 1) матрица - исходная цепь изучаемой ДНК, по образцу которой и происходит синтез новых фрагментов,Идентификация возбудителей инфекционных заболеваний с помощью ПЦР.
В настоящее время эффективность и надежность ПЦР-диагностики заболеваний ни у кого не вызывает сомнений. Сегодня созданы и широко используются в… -ВИЧ-инфекции (признан эталонным методом) -гепатитов В, С, D, E и т.д., в том числе количественная диагностика;Принципы геномной дактилоскопии
Установление родства основано на том, что высокоповторяющиеся фрагменты наследуются. Ребенок получает половину фрагментов от матери и половину от…Тесты для проверки конечного уровня знаний.
1. Участок ДНК, в котором закодирована информация о строении одного белка, называется: А) триплетом; Б) геном;ЛИТЕРАТУРА ОСНОВНАЯ.
1. Спадкові захворювання та природжені вади розвитку в перинатологічній практиці. Запорожан В.М., Сердюк А.М., Бажора Ю.І.та ін.-навч. посібник-К.:Здоров’я,1997. – С.61-71, 139-148.
2. Тект лекции.
Дополнительная.
1. Бочков Н.П. Клиническая генетика.-М.: Медицина,1997.-С.177-182.
ЛИТЕРАТУРА ОСНОВНАЯ.
1. Спадкові захворювання і природжені вади розвитку в перинатологічній практиці (Запорожан В.М., Сердюк А.М., Бажора Ю.1. та iншi ) - навчальний пос1бник.- К.: Здоров’ я.-1997.-С. 78-94, 132-137, 228-241.
2. Резник Б. Я., Запорожан В.Н., Минков И.П. Врожденные пороки развития у детей. Одесса.-1994.-С.323-358.
Дополнительная литература.
1. Бочков Н.П. Клиническая генетика.- М.:Медицина.-1997.-288с.
2. Бердышев Т.Д., Криворучко И.Ф. Медицинская генетика.-К.: Вища школа.-1990.-336с.
3. Наследственные синдромы и медико-генетическое консультирование (Козлова С. И., Демикова Н.С., Семанова Е., Блинникова О.Е.) М.: Медицина, 1996.-416с.
4. Тератология человека. Руководство для врачей. (Под ред. Г.И.Лазюка). - М.:Медицина,1991.-460с.
5. Захаров А.Ф., Бенюш В.А., Кулешов Н.П. и др. Хромосомы человека. (Атлас).- М.:Медицина,1982.-С.8-13, 44-51.
– Конец работы –
Используемые теги: курс, клинической, генетики0.061
Если Вам нужно дополнительный материал на эту тему, или Вы не нашли то, что искали, рекомендуем воспользоваться поиском по нашей базе работ: Курс клинической генетики
Что будем делать с полученным материалом:
Если этот материал оказался полезным для Вас, Вы можете сохранить его на свою страничку в социальных сетях:
| Твитнуть |
Хотите получать на электронную почту самые свежие новости?

Подпишитесь на Нашу рассылку
Реклама





Информация в виде рефератов, конспектов, лекций, курсовых и дипломных работ имеют своего автора, которому принадлежат права. Поэтому, прежде чем использовать какую либо информацию с этого сайта, убедитесь, что этим Вы не нарушаете чье либо право.
© copyright 1999 - 2024 allRefs.net. Все права защищены. Страница сгенерирована за: 0.15 сек.
Новости и инфо для студентов